Claim of Function – Ursprünge von Viren und ihr vermeintlicher Funktionsgewinn
Von MARTIN NEIL and JONATHAN ENGLER
Quelle: wherearethenumbers.substack.com
„Ich habe lieber Fragen, die nicht beantwortet werden können, als Antworten, die nicht in Frage gestellt werden können.“ – Richard P. Feynman
Zusammenfassung
Eine gründliche Überprüfung der verfügbaren Evidenz deutet darauf hin, dass das Auftreten eines neuartigen, manipulierten Virus die unwahrscheinlichste Erklärung für das als “Corona-Pandemie” bekannte Ereignis ist.
Bemerkenswert:
- Die Entdeckung „neuartiger“ Viren hängt davon ab, wie entschlossen wir sind, sie zu finden – je mehr wir suchen, desto mehr finden wir, was darauf hindeutet, dass die Zuschreibung der Neuartigkeit eines Virus eher das Ergebnis eines politisierten Prozesses ist, als dass sie auf einer objektiven Analyse seiner Eigenschaften beruht.
- Die Merkmale von SARS-CoV-2 scheinen nicht so „speziell“ oder „einzigartig“ zu sein wie behauptet.
- Es gibt keine stichhaltigen Beweise dafür, dass die zahlreichen und komplexen biologischen Hürden für die gezielte Entwicklung von Viren, die für den Menschen pathogener oder übertragbarer werden sollen, überwunden worden wären.
- Die Theorie, dass es ein seit langem bestehendes, aber bisher unentdecktes Virus gegeben hätte, das in tierischen (und möglicherweise menschlichen) Reservoiren endemisch gewesen wäre, ist schwer, wenn nicht gar unmöglich zu falsifizieren.
- Es gibt andere Erklärungen für das plötzliche und rasche weltweite Auftreten und die Verbreitung einer bestimmten Gen-Sequenz als die Verbreitung eines neuartigen Virus. Die verfügbaren virologischen und epidemiologischen Beweise stützen weder die Theorie des „Labor Leaks“ noch die des „Fischmarkts“ für den Ursprung des Virus in angemessener Weise.
Wir schlagen daher vor, eher von “Claim-of-Function-” als von “Gain-of-Function-“ Forschung zu sprechen.
Virologische Forschung mit dem Ziel, die Pathogenität zu erhöhen, ist jedoch unethisch und unnötig und sollte daher eingestellt werden. Dies gilt auch dann, wenn wir der Meinung sind, dass die Hypothese, die “Corona-Pandemie“ sei ein iatrogenes1 Phänomen gewesen und nicht durch ein neuartiges und tödliches Virus verursacht worden, durch Beweise untermauert wird. Wenn das Virus tatsächlich eine Funktion hatte, dann die, die Menschheit zu einem dramatischen Akt der Selbstbeschädigung zu verleiten.
Auf die hier vorgestellte Kernthese reagieren viele Menschen mit verschiedenen Formulierungen wie „Fauci und Co. haben es vertuscht, das beweist sicher, dass ein Laborleck die Pandemie verursacht hatte“. Bei dieser Analyse geht es um das Motiv, das bei der Feststellung, ob eine Straftat begangen wurde, zwar beweiskräftig ist, aber eigentlich nur Indiziencharakter hat, da die meisten Gerichtsbarkeiten für einen Schuldspruch direktere Beweise verlangen. In diesem Artikel geht es nicht um das Motiv, sondern um die wichtigere Frage: „Welche Beweise gibt es für die Behauptung, dass die Gain-of-Function-Forschung tatsächlich eine globale Pandemie verursacht hat?” (Eine mögliche Erklärung, wie die Vertuschung des „Lab-Leaks“ mit der „Pandemie-Reaktion“ zusammenhängen könnte, finden Sie hier.)
Ein neuartiger Virus ist nichts Neues
Fields Virology gilt als eines der maßgeblichen Nachschlagewerke im Bereich der Virologie, das sowohl die Virusbiologie als auch die Replikation und die medizinischen Aspekte bestimmter Virusfamilien behandelt. Kapitel 28 von Band 1 dieses Buches wurde von Masters und Perlman verfasst und bietet einige faszinierende Einblicke in unser Wissen über Coronaviren, von denen man annimmt, dass sie den Menschen infizieren, einschließlich:
- HCoV-NL63 und HCoV-HKU1 wurden erst vor kurzem, in der Post-SARS-Ära (2002), entdeckt, obwohl beide weltweit verbreitet sind und schon seit langem im Umlauf sind.
- Vier bekannte Coronaviren – HCoV-OC43, HCoV-229E, HCoV-NL63 und HCoV-HKU1 – sind in menschlichen Populationen endemisch. Die ersten beiden verursachen vermutlich bis zu 30 % aller Infektionen der oberen Atemwege.
- HCoV-NL63 und HCoV-HKU1 sind weltweit verbreitet und verursachen bis zu 10 % der Atemwegsinfektionen.
- Anfängliche Berichte nach seiner „Entdeckung“ ließen vermuten, dass HCoV-NL63 mit schweren Atemwegserkrankungen in Verbindung gebracht wurde; spätere bevölkerungsbezogene Studien zeigten jedoch, dass die meisten Patienten eine leichte Erkrankung entwickelten, ähnlich wie diejenigen, die mit HCoV-229E oder HCoV-OC43 infiziert waren.
- Auch, HECV-4408 Das Coronavirus wurde erstmals 1988 in Deutschland nachgewiesen und wird mit akuter Diarrhöe beim Menschen in Verbindung gebracht und ist (wahrscheinlich) mit Rindercoronaviren verwandt. Die Literatur zu diesem Virus ist jedoch äußerst spärlich, und es scheint kein aktives Forschungsinteresse an diesem Virus zu bestehen.
Pyrc et al. berichten, dass HCoV-OC43 und HCoV-229E Mitte der 1960er Jahre entdeckt wurden und seitdem weltweit verbreitet sind. Es ist zwar nicht ganz klar, wie die Prävalenz festgestellt wurde, doch scheint man davon auszugehen, dass sie bereits endemisch waren und nicht neu, entweder in einem Labor oder durch Zoonose entstanden. HCoV-NL63 und HCoV-HKU1 waren Coronaviren, die der Wissenschaft bis vor relativ kurzer Zeit unbekannt waren, obwohl sie weltweit einen hohen Anteil an Atemwegsinfektionen verursachen und dies wahrscheinlich schon seit Jahrhunderten, wenn nicht sogar seit Anbeginn der Zeit tun (wie später beschrieben). Ganze ein Viertel der Coronavirus-Infektionen waren auf diese der Wissenschaft bis dahin völlig unbekannten Coronaviren zurückzuführen. Wie wir später noch erläutern werden, wurde eines dieser Viren bei seiner Entdeckung mit schweren Atemwegserkrankungen in Verbindung gebracht, was sich jedoch nach einer sorgfältigen Analyse auf Bevölkerungsebene als falsch erwies.
Es gibt auffallende Ähnlichkeiten zwischen den Coronaviren HCoV-NL63 und HCoV-HKU1 und SARS-CoV-2. Sie wurden alle neu entdeckt, zwei von ihnen wurden mit schweren Erkrankungen in Verbindung gebracht, und bei zwei von ihnen wurde der Schweregrad deutlich herabgestuft, nachdem die Populationsdaten analysiert worden waren.
Was jedoch den Ursprung von SARS-CoV-2 betrifft, so haben wir die Wahl zwischen zwei konkurrierenden Geschichten: Entweder war es das Produkt eines Laborlecks, oder es entstand durch zoonotische Übertragung von Tieren auf Menschen, und in beiden Fällen geschah dies kurz vor der ersten Entdeckung. Die dritte Möglichkeit – eine bereits bestehende Endemie – wird kaum als Möglichkeit diskutiert. Eine Analogie zu dieser Situation wäre es, das hochauflösende Hubble-Teleskop einzuschalten und zu erklären, dass die neu entdeckten Exoplaneten (Planeten, die um andere Sterne als unsere Sonne kreisen) in dem Moment oder kurz davor entstanden sind, als das Teleskop eingeschaltet wurde, anstatt davon auszugehen, dass sie schon seit Äonen da waren.
Warum wird bei einigen neu entdeckten Coronaviren (HCoV-NL63 und HCoV-HKU1) davon ausgegangen, dass sie bereits endemisch sind, während man bei anderen davon ausgeht, dass sie völlig neu sind und eine Pandemie auslösen können? Dies sollte eine wichtige wissenschaftliche Frage sein, aber nur wenige haben sich die Mühe gemacht, sie zu stellen, geschweige denn zu versuchen, sie zu beantworten.
Ein interessanter Artikel über die Entdeckung eines neuen Virus, Redondoviridae, im Jahr 2019 stellt die Frage: Wie findet man ein Virus, das völlig unbekannt ist? Sie weisen darauf hin, dass:
„Viren, die am häufigsten vorkommenden biologischen Einheiten auf der Erde, sind eine Geißel der Menschheit und verursachen sowohl chronische Infektionen als auch globale Pandemien, die Millionen von Menschen töten können. Dennoch ist das wahre Ausmaß der Viren, die Menschen infizieren, nach wie vor völlig unbekannt. Einige neu entdeckte Viren werden erst durch das plötzliche Auftreten einer neuen Krankheit erkannt, wie z. B. SARS im Jahr 2003 oder auch HIV/AIDS in den frühen 1980er Jahren. Neue Techniken ermöglichen es den Wissenschaftlern nun, Viren durch die direkte Untersuchung von RNA- oder DNA-Sequenzen im genetischen Material des Menschen zu identifizieren, was den Nachweis ganzer Viruspopulationen – des so genannten Viroms – ermöglicht, einschließlich solcher, die möglicherweise keine akut erkennbare Krankheit verursachen. Die Identifizierung neuartiger Viren ist jedoch schwierig, da ihre genetischen Sequenzen möglicherweise wenig mit bereits bekannten Virusgenomen gemeinsam haben, die in Referenzdatenbanken verfügbar sind.“
Sie berichten weiter, dass Forscher der Universität von Pennsylvania eine bisher unbekannte Virusfamilie identifiziert haben, die sich als das zweithäufigste DNA-Virus in menschlichen Lungen- und Mundproben entpuppt, wo es mit schweren kritischen Erkrankungen und Zahnfleischerkrankungen in Verbindung gebracht wird!
Sind neue Viren also wirklich so neu? Wie wir gesehen haben, wurden seit den 1980er Jahren mindestens sechs „neue“ Coronaviren entdeckt (MERS-CoV, SARS, SARS-CoV-2, HCoV-NL63, HCoV-HKU1 und HECV-4408), also durchschnittlich eines alle sieben Jahre. Wie können wir ausschließen, dass sie nicht doppelt so viele Viren gefunden hätten, wenn sie ihre Bemühungen verdoppelt hätten, diese scheinbar neuen Viren zu finden?
MERS-CoV und SARS als historischer Beleg für den Trugschluss einer einzigen Ursache
In einem Aufsatz von 2014 MacIntyre stellte MacIntyre fest, dass das Middle East Respiratory Syndrome Coronavirus (MERS-CoV) eine neu aufgetretene Infektion ist, die Menschen auf der Arabischen Halbinsel, in Europa und Nordafrika befällt. Die meisten MERS-CoV-Fälle wurden mit Krankenhausausbrüchen in Jordanien, KSA, den Vereinigten Arabischen Emiraten und Frankreich in Verbindung gebracht. Die klinischen Merkmale dieser Cluster waren recht unterschiedlich, wobei beim ersten Ausbruch in Jordanien vor allem Nierenversagen auftrat, das in anderen Clustern weniger oder gar nicht vorkommt und ein ungewöhnliches Merkmal einer durch ein Atemwegsvirus verursachten Krankheit wäre1.
Ein einziger Infektionserreger sollte sich nicht auf so unterschiedliche Weise manifestieren. Nach einem altmodischen Krankheitsmodell würde MERS-CoV als eigenständige Krankheiten eingestuft werden, und nur aufgrund der neumodischen Betrachtungsweise von Sequenzen und der Tatsache, dass eine Sequenz nachgewiesen (oder vermutet) wird, wird angenommen, dass es eine einzige Ursache geben muss – „das Virus“. Diese Annahme ist jedoch ein Beispiel für den „Trugschluss der einzigen Ursache“ und hat keine klinische Gültigkeit; sie ist ein Täuschung.
Der Ursprung und die Persistenz der Infektion beim Menschen sind nach wie vor unbekannt, obwohl innerhalb von zwei Jahren 681 Menschen infiziert wurden und 204 starben. Die Fälle konzentrierten sich auf den Nahen Osten, und die Infektion scheint sich nicht über diese Region hinaus verbreitet zu haben. Bemerkenswert ist die Aussage des Autors:
„Als die beobachteten Daten an verschiedene Krankheitsmuster angepasst wurden, passten die Merkmale von MERS-CoV besser zu einem in unregelmäßigen Abständen wiederkehrendem Muster, das entweder auf eine absichtliche Freisetzung oder eine tierische Quelle hindeutet. Es gibt viele Diskrepanzen in der beobachteten Epidemiologie von MERS-CoV, die besser zu einem sporadischen als zu einem epidemischen Muster passen.“
Obwohl das Virus bei Fledermäusen und Kamelen gefunden worden war, wurde vermutet, dass sich die meisten Menschen asymptomatisch mit dem Virus ansteckten. Kamele galten als die wahrscheinlichste Quelle, obwohl es keine besonders überzeugenden Beweise für eine Übertragung auf den Menschen durch Kamele gab, außer dass das Virus bei einigen Kamelen gefunden worden war2. Größere Ausbrüche gab es in Südkorea im Jahr 2015 und in Saudi-Arabien im Jahr 2018.
Darüber hinaus gibt es immer wieder Spekulationen über Bioterrorismus (ohne jeden Beweis):
„Schließlich rechtfertigt die abweichende Epidemiologie eine kritische Analyse aller möglichen Erklärungen und die Einbeziehung aller Beteiligten in die Biosicherheit, und eine absichtliche Freisetzung muss ernsthaft in Betracht gezogen und zumindest als Möglichkeit anerkannt werden.
Gardener und MacIntyre bestreiten, dass das Muster von MERS-CoV überhaupt als sporadisch, geschweige denn als epidemisch angesehen werden sollte, denn obwohl es während der Hadsch- und Umrah-Pilgerreisen nach Saudi-Arabien im Umlauf war, also unter angeblich perfekten Bedingungen für eine Epidemie, hat es sich nicht sichtbar ausgebreitet. Sie stellen fest:
„Zu den mit MERS-CoV assoziierten Risikofaktoren gehören das männliche Geschlecht, Grunderkrankungen, Immunsuppression und Krankenhausaufenthalte.”
Sie sagen auch, dass MERS-CoV in erster Linie eine nosokomiale (im Krankenhaus erworbene) Infektion war, die mit bereits im Krankenhaus befindlichen Personen in Verbindung gebracht wurde. Sie sagen, dass auch SARS überwiegend eine nosokomiale Infektion war.
In einem Vergleich zwischen SARS-CoV-2 und MERS-CoV Khalid et al. berichten das:
„Der Verlauf nach der Intubation war in beiden Gruppen ähnlich. Die Patienten beider Gruppen wurden länger mechanisch beatmet und erhielten mehrheitlich Paralytika, Dialyse und vasopressorische Mittel. Die Atmungs- und Beatmungsparameter nach der Intubation …. und ihr Verlauf über 3 Wochen waren ähnlich. Die Sterblichkeitsrate auf der Intensivstation (53 % vs. 64 %) und im Krankenhaus (59 % vs. 64 %) war bei COVID-19- und MERS-Patienten sehr hoch.“
Abid et al. bestätigten, dass die MERS-CoV-Fälle durch bakterielle und Pilzinfektionen erschwert wurden. Die Hälfte der Patienten wies eine Sauerstoffsättigung von weniger als 90 % auf, und die meisten Patienten starben nach längerer Beatmung. Antibiotika wurden bei der Einlieferung ins Krankenhaus verabreicht (was möglicherweise zu spät war, hier).
SARS wurde erstmals 2002 mit den ersten bekannten Fällen entdeckt und soll den Ausbruch von 2002 bis 2004 verursacht haben, hauptsächlich in China (Andersen et al.). Wikipedia bietet einen guten Überblick über das, was über SARS bekannt ist, wobei der interessanteste Unterschied zu MERS-CoV darin besteht, dass die WHO erklärte, dass SARS als akutes respiratorisches Syndrom im Jahr 2003 aufgrund von Eindämmungsmaßnahmen (die auf Maßnahmen der öffentlichen Gesundheit zurückzuführen sind) verschwunden ist. In den Jahren 2003 und 2004 breitete sich das Virus jedoch trotz vier Laborunfällen nicht aus (hier), und es wird behauptet, dass es eine anhaltende zoonotische Bedrohung darstellt.
Kommt Ihnen das bekannt vor?
Man geht davon aus, dass es eine Infektion gibt und dass sie sich weltweit ausbreiten könnte, auch wenn die Ausbreitungsmuster den derzeit maßgeblichen epidemiologischen Mechanismen widersprechen.
- Die Infektion wird hauptsächlich im Krankenhaus erworben, wobei die einzige in Betracht zu ziehende Annahme darin besteht, dass ein neues Virus erst vor kurzem in der Natur aufgetaucht sei, oder es wird vermutet, dass es vom Menschen erzeugt wurde – die Möglichkeit einer bereits bestehenden Endemie wird ignoriert.
- Die vermeintlich hohe Sterblichkeitsrate bei bereits stationär behandelten Fällen wird auf das Virus zurückgeführt und ist unabhängig von der Grunderkrankung der Patienten.
- Die Möglichkeit einer asymptomatischen Ausbreitung wird einfach vorausgesetzt, ohne dass es dafür Beweise gibt. Dadurch wird die Möglichkeit einer Pandemie aufgebauscht, obwohl die alternative und einfache Schlussfolgerung lautet, dass eine unsichtbare Ausbreitung nicht so tödlich sein kann.
- Sowohl bei MERS-CoV als auch bei SARS sind die Symptome unspezifisch und verursachen grippeähnliche Beschwerden. Nichts war diagnostisch. Es hieß auch, dass Röntgenaufnahmen diagnostisch für SARS seien, aber wie wir in unserem Artikel über Spikeopathie keine radiologischen Anzeichen zwischen dieser und einer anderen Viruserkrankung der Atemwege unterscheiden konnten.
- Es ist bekannt, dass beide zu einer bakteriellen Lungenentzündung führen können.
- Wikipedia heißt es, dass 72 % der Menschen wegen MERS-CoV mechanisch beatmet werden mussten und die Beatmung zur Behandlung von SARS eingesetzt wurde.
- Sowohl bei SARS als auch bei MERS-CoV wurde von einer Antibiotikabehandlung abgeraten, auf Grund der WHO Richtlinien.
- Beide wurden durch PCR diagnostiziert, aber die WHO und das CDC haben die Diagnose auf bloßen Verdacht hin empfohlen. CDC (eine bloße Häufung unerklärlicher Lungenentzündungen würde ausreichen).
Auch hier sehen wir wieder den Trugschluss, dass es nur eine einzige Ursache gibt, und werden mit diesen höchst spekulativen Erklärungen über die Krankheiten konfrontiert, die angeblich durch diese vergangenen Epidemien verursacht wurden, und zwar durch Viren und nur durch Viren.
Der springende Punkt ist, dass die Mortalität und Morbidität von SARS und MERS-CoV höchstwahrscheinlich stark überschätzt werden, was auf die verwirrenden Auswirkungen unangemessener medizinischer Behandlungen und Diagnosen zurückzuführen ist, die auf der Annahme einer hohen Prävalenz und einer langen Reisegeschichte basieren. Daher wird die offensichtliche Virulenz und Pathogenität neu entdeckter Viren offenbar regelmäßig kurz nach ihrer Entdeckung stark überschätzt, nur um später entweder massiv herabgestuft zu werden (HCoV-NL63) oder die Fiktion ihrer Schwere unangefochten zu bleiben (SARS-CoV-2, SARS, MERS-CoV). Dieses Schema wiederholt sich immer wieder, wie jüngst bei H5N1 (hier).
Würden wir ein neues Virus erkennen, wenn wir es sehen?
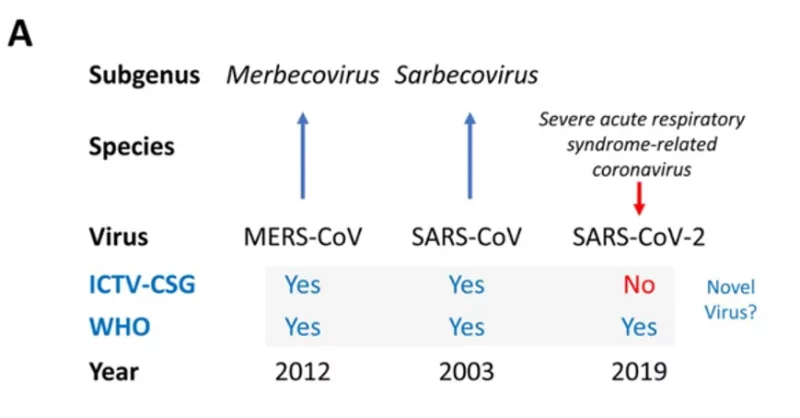
Wenn uns gesagt wird, dass wir SARS-CoV-2 als neuartig akzeptieren sollen, sollte diese Akzeptanz davon abhängen, wie robust das Verfahren war, mit dem das Virus als „neuartig“ bezeichnet wurde. In diesem Zusammenhang ist es sinnvoll, den Entscheidungsprozess zu untersuchen, der dieser Entscheidung zugrunde liegt (Engler).
Im Februar 2020 bewertete das International Committee on Taxonomy of Viruses (ICTV-CSG) der Coronavirus Study Group (CSG), das für die Entwicklung der offiziellen Klassifizierung von Viren und die Benennung von Taxa (Taxonomie) der Familie der Coronaviridae zuständig ist, die Neuartigkeit des menschlichen Erregers mit der vorläufigen Bezeichnung 2019-nCoV (Vorabdruck auf biorxiv Gorbalenya et al., Perlman und Drosten sind Mitautoren).
Das Virus wurde vorübergehend als 2019 novel coronavirus, 2019-nCoV, bezeichnet und auf der Grundlage der Empfehlungen der CSG in SARS-CoV-2 umbenannt.
So brachten sie die Herausforderungen bei der Entscheidung über die Neuheit zum Ausdruck:
„Der Begriff „neuartig“ kann sich auf die Krankheit (oder das Spektrum der klinischen Manifestationen) beziehen, die bei Menschen, die mit diesem speziellen Virus infiziert sind, verursacht wird, die jedoch erst im Entstehen begriffen ist und weitere Studien erfordert. Der Begriff „neuartig“ im Namen von 2019-nCoV kann sich auch auf eine unvollständige Übereinstimmung zwischen den Genomen dieses und anderer (bisher bekannter) Coronaviren beziehen, wenn letzteres als geeignetes Kriterium für die Definition von „Neuartigkeit“ angesehen würde. Virologen sind sich jedoch einig, dass weder die Krankheit noch das Wirtsspektrum zur zuverlässigen Bestimmung der Neuartigkeit (oder Identität) des Virus herangezogen werden können, da nur wenige Genomveränderungen ein tödliches Virus abschwächen oder einen Wirtswechsel verursachen können.Ebenso wissen wir, dass RNA-Viren als ein Schwarm von sich gemeinsam entwickelnden, eng verwandten Entitäten (Varianten einer bestimmten Sequenz, Haplotypen), den so genannten Quasi-Spezies, fortbestehen. Ihre Genomsequenz ist eine konsensbasierte Momentaufnahme einer sich ständig weiterentwickelnden kooperativen Population in vivo und kann innerhalb einer einzelnen infizierten Person und im Laufe der Zeit bei einem Ausbruch variieren.
Würde das Kriterium der strikten Übereinstimmung auf RNA-Viren angewandt, so würde jedes Virus mit einem sequenzierten Genom als neuartiges Virus eingestuft, was dieses Kriterium wenig aussagekräftig macht. Um das potenzielle Problem zu umgehen, können Virologen stattdessen zwei Viren mit nicht identischen, aber ähnlichen Genomsequenzen als Varianten desselben Virus betrachten; dies wirft sofort die Frage auf, wie viel Unterschied groß genug ist, um das Kandidatenvirus als neu oder anders zu erkennen. Diese Frage wird in der Praxis am besten dadurch beantwortet, dass man den Grad der Verwandtschaft des Kandidatenvirus mit bereits bekannten Viren desselben Wirts oder mit etablierten monophyletischen Gruppen von Viren, die oft als Genotypen oder Kladen bekannt sind und Viren verschiedener Wirte umfassen können oder auch nicht, bewertet.“
Die Neuartigkeit hängt also von einer Genomsequenz ab, die nur eine Momentaufnahme eines sich ständig weiterentwickelnden dynamischen Schwarms verwandter Entitäten ist, und daher ist die Entscheidung darüber, was eine Neuartigkeit darstellt, komplex und eher einer Beurteilung als einer objektiven Analyse geschuldet. Außerdem hängt sie sowohl von der Krankheit oder den klinischen Symptomen als auch von der Vollständigkeit oder Unvollständigkeit der Vergleiche mit anderen Viren ab. Dies stellt ein wahres Sammelsurium von Verwechslungen und Verwechslungen von Ursache und Wirkung dar und definiert keineswegs eine „Sache“ an sich.
Würde man das strikte Neuheitskriterium auf RNA-Viren anwenden, so wäre jedes Virus mit einem sequenzierten Genom ein neuartiges Virus. Daher können sie nicht als isolierte Singles mit absolut einzigartigen Merkmalen betrachtet werden, sondern eher als sich gegenseitig überlappende Familien von Individuen mit gemeinsamen Merkmalen, wodurch die Idee der Neuheit in einem absoluten Sinne völlig überflüssig wird. Das ICTV wendet daher eine Taxonomie auf Viren an, die fünf hierarchisch angeordnete Ränge vorsieht: Ordnung, Familie, Unterfamilie, Gattung und Art (in aufsteigender Reihenfolge der Ähnlichkeit zwischen den Viren). Kritiker dieses Ansatzes argumentieren, dass die genetische Vielfalt nur zuverlässig mathematisch ausgedrückt werden kann, also über die paarweisen Abstände zwischen ihnen und Überschneidungswahrscheinlichkeiten bei der genetischen Divergenz. Oder anders ausgedrückt: Es kann keine Taxonomie (Klassifikationsschema) in einem Schwarm geben.
Als die Studie in Nature Mikrobiology veröffentlicht wurde, waren alle Hinweise auf die Schwierigkeiten und die Rolle von Expertenmeinungen bei der Vergabe des Namens SARS-CoV-2 gestrichen worden, so dass der Eindruck entstand, die Entscheidung sei alternativlos und in keiner Weise umstritten. Wäre der Name 2019-nCoV geblieben, hätte man es wahrscheinlich als ein weiteres Erkältungsvirus betrachtet, über das man sich keine Sorgen machen muss.
Im Vordruck bezeichnet die ICTV-CSG SARS-CoV-2 eindeutig als nicht neuartiges Virus und zeigt damit, dass sie zu einer anderen Entscheidung gekommen ist als die WHO (und zwar mit roter Tinte, um dies deutlich zu machen):
Es gibt einige Hinweise auf mögliche Meinungsverschiedenheiten zwischen der ICTV-CSG und der WHO (dieser Absatz wurde nicht in das von Nature veröffentlichte Papier aufgenommen):
„Im Gegensatz zu SARS wurde der Name SARS-CoV-2 NICHT vom Namen der SARS-Krankheit abgeleitet und sollte keinesfalls dazu verwendet werden, den Namen der durch SARS-CoV-2 beim Menschen verursachten Krankheit (oder des Krankheitsspektrums) vorzugeben, der von der WHO festgelegt werden wird. Die verfügbaren, jedoch begrenzten epidemiologischen und klinischen Daten zu SARS-CoV-2 deuten darauf hin, dass sich das Krankheitsspektrum und die Übertragungswege dieses Virus und von SARS unterscheiden könnten. Auch die diagnostischen Methoden zur Bestätigung von SARS-CoV-2-Infektionen sind nicht identisch mit denen von SARS. Dies spiegelt sich in den spezifischen Empfehlungen für Ärzte, Beschäftigte im Gesundheitswesen und Labordiagnostiker für SARS-CoV-2/2019-nCoV wider (z. B. in den WHO Leitlinien für 2019-nCoV). Durch die Entkopplung der Bezeichnungskonventionen für Coronaviren und die Krankheiten, die sie bei Mensch und Tier verursachen können, möchten wir der WHO helfen, Krankheiten auf die am besten geeignete Weise zu benennen (WHO Richtlinien für die Benennung von Krankheiten).“
Sie weisen auch auf die Möglichkeit hin, dass sowohl SARS als auch SARS-CoV-2 verdächtige Ursprünge haben:
„Obwohl SARS-CoV-2 KEIN Nachkomme von SARS ist und die Einschleppung beider Viren in den Menschen wahrscheinlich durch unbekannte externe Faktoren begünstigt wurde, sind sich die beiden Viren genetisch so ähnlich, dass ihre Evolutionsgeschichte und ihre Merkmale gegenseitig aufschlussreich sind.“
Und dann gestehen Sie, dass Meinungsverschiedenheiten über die Neuheit selbst nicht neu sind:
„Da die derzeitige Stichprobe von Viren klein und stark auf Viren von bedeutendem medizinischem und wirtschaftlichem Interesse ausgerichtet ist, variiert die Gruppenzusammensetzung zwischen den verschiedenen Viren enorm, so dass Entscheidungen über die Neuheit gruppenspezifisch sind und von der Wahl spezifischer Kriterien durch die Forscher abhängen. In der Praxis bedeutet dies, dass endgültige Beweise für die Neuartigkeit in einer Gruppe einer Überprüfung in einer anderen Gruppe möglicherweise nicht standhalten.“
Die Vorstellung, dass es verschiedene Arten gibt, die in saubere, sich gegenseitig ausschließende Kategorien fallen, ist fragwürdig. Im Tierreich überschneiden sich die vermeintlich neuen Arten oder Unterarten. Nehmen wir zum Beispiel die Liger – der hybride Nachkomme von Tigern und Löwen. Auch verschiedene Unterarten von Bären kreuzen sich ganz leicht. Das Leben ist ein Kontinuum, und wenn es schon nicht möglich ist, Tiere zu kategorisieren und in eine Taxonomie einzuordnen, sollten wir dann nicht auch bei Viren übervorsichtig sein, bei Organismen, die sich nicht so leicht beobachten lassen und über die wir viel weniger wissen? Wir gehen davon aus, dass sich das ICTV dieser Grenzen der Wissenschaft der Virologie bewusst ist, zumal die Debatten über die Taxonomie von Viren seit ihren Anfängen toben, wobei viele prominente Wissenschaftler innerhalb der Disziplin die Ansicht vertreten, dass absteigende hierarchische Einteilungen auf willkürlichen und monothetischen Annahmen beruhen (siehe Murphy et al. – Einführung in das universelle System der Virustaxonomie).
Die Zuweisung des Wortes “neuartig” und die Verwendung des Namens wirken wie das Ergebnis eines umstrittenen politischen Prozesses oder eines sozialen Konstrukts, das als wissenschaftlich dargestellt wird und bei dem alle Unsicherheiten, Zweifel und Streitigkeiten beseitigt wurden.
Sind Spike-Proteine, Inserts und Furin-Spaltstellen gefährlich oder neuartig?
Es wurde viel über die Tatsache berichtet, dass SARS-CoV-2 sowohl ein Spike-Protein (das S-Protein) als auch eine Furin-Spaltstelle (FCS) enthält. Dies hat zu großer Besorgnis und Kontroversen geführt. Einige behaupten, dass das Spike-Protein besonders gefährlich ist und dass die „einzigartige“ Furin-Spaltstelle in diesem Spike-Protein für die hohe Infektiosität und Übertragbarkeit verantwortlich ist.
Aber sind sie an sich schon gefährlich und neuartig?
Xia et al. sagen in der Fachzeitung Nature:
„Die rasche Ausbreitung von SARS-CoV-2 …..einem neuartigen Betacoronavirus der Linie B… hat eine globale Pandemie von Coronavirus-Erkrankungen (COVID-19) verursacht. Es wurde spekuliert, dass RRAR, eine einzigartige Furin-ähnliche Spaltstelle (FCS) im Spike-Protein (S), die bei anderen Betacoronaviren der Linie B, wie z. B. SARS-CoV, fehlt, für die hohe Infektiosität und Übertragbarkeit des Virus verantwortlich ist. …
Daher wurde spekuliert, dass dieses einzigartige FCS einen Funktionsgewinn darstellt, der es SARS-CoV-2 im Vergleich zu anderen Betacoronaviren der Linie B erleichtert, in die Wirtszelle einzudringen, um sich dort zu infizieren und sich so effizient in der menschlichen Bevölkerung zu verbreiten.“
Seine Ausstülpung wird beschrieben als seltsam und wird als „durch Experimentieren entstanden“ beschrieben. Es wurden spekulative Behauptungen aufgestellt, dass die FCS Fibrose hervorruft. Die angebliche Zerbrechlichkeit des Spike-Proteins gegenüber Antikörpern wirft Fragen nach dem Ursprung auf.
Dieser Bericht von Liu et al. identifiziert:
„….248 andere CoVs mit 86 verschiedenen Furin-Spaltstellen, die seit 1954 in 24 Tierwirten in 28 Ländern nachgewiesen wurden.
….Neben MERS-CoV und SARS-CoV-2 haben auch zwei der fünf anderen CoVs, von denen bekannt ist, dass sie Menschen infizieren (HCoV-OC43 und HCoV-HKU1), Furin-Spaltstellen. Darüber hinaus verfügt das humane enterische Coronavirus (HECV-4408) über eine Furin-Spaltstelle….“
Die folgende Abbildung zeigt die phylogenetischen Beziehungen der Coronavirus-Spike-Proteine mit Furin-Spaltstellen.
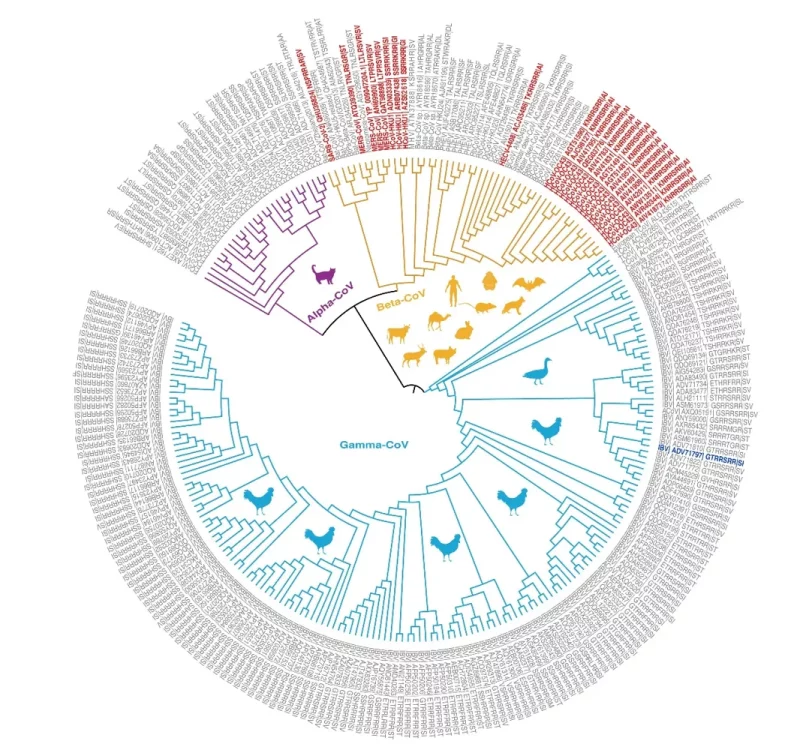
Phylogenetische Beziehungen von Coronavirus-Spike-Proteinen mit Furin-Spaltstellen.
Das Papier von Ambati et al. argumentiert, dass die Zusammensetzung des SARS-CoV-2 FCS sehr ungewöhnlich ist, aber „ungewöhnlich“ bedeutet wenig ohne einen Vergleich mit dem Typischen. Sie sagen:
„…das Vorhandensein der 19-Nukleotid langen RNA-Sequenz einschließlich der FCS mit 100%iger Identität zum reversen Komplement der MSH3 mRNA ist höchst ungewöhnlich und erfordert weitere Untersuchungen.“
Sie durchsuchten eine Datenbank mit 24.712 genomischen Sequenzen und berechneten eine Koinzidenzwahrscheinlichkeit von 3,21 × 10−11 für die SARS-CoV-2-Furinspaltsequenz, woraus sie schlossen, dass die FCS vom Menschen hergestellt wurde.
Dubuy und Lachuer bestritten diese Behauptung, indem sie die Wahrscheinlichkeit des Zusammentreffens anderer Viren mit denselben Merkmalen untersuchten. Auf der Grundlage einer sorgfältigen probabilistischen Analyse, bei der berücksichtigt wurde, dass die Sequenzen nicht unabhängig sind, dass die Sequenzen unterschiedlich lang sind und andere Faktoren berücksichtigt wurden, kamen sie zu einer Wahrscheinlichkeit von 0,0037 – eine Größenordnung höher als die von Ambati et al. berechnete Wahrscheinlichkeit.
Man beachte, dass die Hauptannahme in der Arbeit von Dubuy und Lachuer darin besteht, dass Viren dieselben Merkmale aufweisen könnten, vielleicht aufgrund einer parallelen Evolution, aber auch aufgrund einer Koevolution, bei der eine Rekombination zwischen verschiedenen Virusspezies stattfindet, was genau das ist, was man in einem Virenschwarm erwarten könnte, wie das ICTV bestätigt.
Daher könnte das FCS auf natürliche Weise entstanden sein und war vielleicht einfach ein neu entdecktes Virus, das in der Umwelt bereits weit verbreitet war. Daher ist weder am Spike-Protein noch am FCS wirklich viel Neues zu finden. Masters und Perlman weisen darauf hin:
- Bei vielen Beta- und Gamma-Coronaviren (z. B. Maushepatitisvirus, bovines Coronavirus und infektiöses Bronchitisvirus) wird das S-Protein durch eine Furin-ähnliche Protease der Wirtszelle teilweise oder vollständig in zwei Polypeptide gespalten…., die ungefähr gleich groß sind.
- Die derzeit verfügbaren strukturellen und biochemischen Beweise stimmen gut mit einem frühen Vorschlag überein, wonach S (spike) funktionell dem Influenza-HA-Protein ähnelt.
Auch die Tatsache, dass das Spike an menschliche ACE2-Rezeptoren bindet, wird häufig erwähnt. Nochmals zurück zu Masters und Perlman:
„Der Rezeptor für SARS – das Angiotensin-Converting Enzyme 2 (ACE2) – wurde nach der Isolierung des Virus in bemerkenswert kurzer Zeit entdeckt.
……
ACE2 dient auch als Rezeptor für das Alphacoronavirus HCoV-NL63, und der entsprechende Strukturkomplex für dieses Virus zeigt, dass die RBD (rezeptorbindende Domäne) von HCoV-NL63 und die RBD von SARS an dieselben Motive binden“.
Wir haben es hier also mit einem anderen Coronavirus zu tun, das früher als „tödlich“ bezeichnet wurde, das ebenfalls den ACE2-Rezeptor nutzt, aber (im Wesentlichen) eine leichte Atemwegserkrankung verursacht.
Es ist zu beachten, dass ACE2 in weniger als 1 % der menschlichen Lungenzellen exprimiert wird (hier), wobei es am häufigsten in den Alveolarzellen vom Typ II vorkommt, die ihrerseits nur 5 % der Alveolarfläche ausmachen, während die anderen 95 % der Fläche von Plattenepithelzellen bedeckt sind. Daher ist es fraglich, ob die menschliche Lungenfunktion entscheidend von ACE2 abhängt (wie es bei Mäusen der Fall sein könnte, die speziell für menschliche ACE2-Rezeptoren gezüchtet wurden).
Die Vorveröffentlichung und anschließende Rücknahme einer Arbeit von Pradham et al. wurde sehr kontrovers diskutiert. Pradham et al. behaupten ein Glykoprotein namens gp120 in den Spikes des 2019-nCoV-Virus gefunden zu haben. Man geht davon aus, dass dieses Protein nur für das HIV-Virus spezifisch ist, was darauf hindeutet, dass das Virus vom Menschen hergestellt sein muss. Der Nobelpreisträger von 2008, Luc Antoine Montagnier, replizierte den Befund und unabhängige Forscher haben ihn seitdem bestätigt (Rose). Zufälligerweise oder auch nicht, arbeitete ein anderes, von Deutschland finanziertes Labor in Wuhan an einem HIV-Impfstoff, der die Quelle des Ausbruchs gewesen sein könnte (Kogon).
Ist gp120 einzigartig für das HIV-Virus? Das Simian-Immunschwäche-Virus (SIV) ist als Vorläufer von HIV identifiziert worden, und bei SIV ist es auf dem SIV-Spike-Protein vorhanden. Da Rekombinationen zwischen HIV oder SIV und Coronaviren nicht auszuschließen sind, kann nicht ausgeschlossen werden, dass SIV die Quelle für das Vorhandensein von gp120 im SARS-CoV-2-Spike-Protein ist. Wir haben zwar keine Ahnung, wie viel HIV und SIV „da draußen“ sind, und dieser Gedanke hat keinerlei Beachtung gefunden, aber das Fehlen von Beweisen kann nicht als Beweis für das Fehlen von Beweisen gewertet werden3 (beachten Sie, dass eine asymptomatische HIV-Infektion mehr als 10 Jahre dauern kann und die Latenzzeit der SIV-Infektion weitgehend unerforscht ist (Anmerkung der Redaktion: Das ist zumindest das herrschende Narrativ. Dem widersprechen verschiedene Veröffentlichungen von OVALmedia, z.B. diese Artikel von Johannes Kreis hier und hier)
Daraus können wir schließen:
- Spikes bei Coronaviren sind nichts Besonderes.
- Spike-Proteine ähneln den Proteinen anderer Viren, einschließlich Influenza, oder haben sie mit ihnen gemeinsam.
- FCS sind weit verbreitet. Aber die überwiegende Mehrheit der (bisher) identifizierten 248 wurde nicht annähernd so genau untersucht wie SARS-CoV-2.
- ACE-2-Rezeptoren werden auch von einem anderen Coronavirus, HCoV-NL63, genutzt, das nicht als tödlich gilt.
- Argumente, dass „ungewöhnliche Einschübe“ vom Menschen stammen müssen, könnten durch eine parallele Evolution oder Koevolution zwischen Viren „im Schwarm“ widerlegt werden.
Daher sehen wir keinen Grund zu der Annahme, dass das Spike-Protein oder FCS bei SARS-CoV-2 Anlass zu besonderer und einzigartiger Besorgnis geben sollte. Selbst diese „ungewöhnlichen Einschübe“, die das Virus angeblich bemerkenswert machen, könnten in der Tat nicht bemerkenswert sein, da es viel wahrscheinlicher (wenn auch nicht sicher) erscheint, dass andere Viren die gleichen Merkmale durch natürliche Koevolution zwischen Viren enthalten.
Neuartigkeit der SARS-CoV-2 Furin-Spaltstelle
Ein Großteil des Streits über die Entstehungshypothese für SARS-CoV-2 dreht sich um die Neuartigkeit bestimmter Merkmale des Virus und darum, ob diese den in der Natur vorkommenden oder den vom Menschen geschaffenen Viren ähneln (und mit den eingereichten Forschungsvorschlägen übereinstimmen).
Einen Überblick über die Hinweise, die die Hypothese stützen, dass SARS-CoV-2 in einem Labor künstlich hergestellt wurde, finden Sie hier. Ein unterhaltsamer und fundierter Artikel, der die Beobachtungsdaten debattiert, die eine der beiden Hypothesen unterstützen, wurde verfasst von PANDA.
Die Labortheorie stützt sich zum Teil auf die Tatsache, dass bisher keine Fledermauskolonien gefunden wurden, die mit genau diesem Virus infiziert waren, so dass das Virus in der Natur nie zuvor existiert hatte. Die Vermutung, dass es von Menschenhand geschaffen wurde, wurde dann durch die Enthüllung gestützt, die durch FOIA4 die Existenz des DEFUSE-Projektvorschlags, eines Antrags an die DARPA (eine Forschungsbehörde des Pentagon) auf Gewährung eines Zuschusses zur Verbesserung von SARS-ähnlichen Fledermausviren (wir werden später sehen, dass dies kein einmaliges Vorhaben war).
Der Gedanke an eine undichte Stelle im Labor wird als schockierend dargestellt, aber Nass hat zwischen den Jahren 2000 und 2021 309 im Labor erworbene Infektionen und 16 Entweichungen dokumentiert, darunter einige, die mehrere Todesfälle verursachten. Sie verursachten jedoch keine Pandemien.
Das Preprint-Papier von Bruttel et al. argumentiert, dass SARS-CoV-2 aufgrund des „Endonuklease-Fingerabdrucks“ (Endonuklease-Erkennungsstellen BsmBI/BsaI), der in SARS-CoV-2 gefunden wurde, künstlich hergestellt wurde. BsmBI/BsaI werden üblicherweise für das molekulare Klonen verwendet. Sie sagen:
„Wir haben festgestellt, dass SARS-CoV (das ursprüngliche SARS) den für synthetische Viren typischen Restriktionsstellen-Fingerabdruck aufweist. Der synthetische Fingerabdruck von SARS-CoV-2 ist bei Coronaviren in freier Wildbahn anomal und bei im Labor hergestellten Viren üblich…
….Die Restriktionskarte von SARS-CoV-2 stimmt mit vielen zuvor gemeldeten synthetischen Coronavirus-Genomen überein, erfüllt alle Kriterien, die für ein effizientes reverses genetisches System erforderlich sind, unterscheidet sich von seinen nächsten Verwandten durch eine deutlich höhere Rate an synonymen Mutationen in diesen synthetisch aussehenden Erkennungsstellen und hat einen synthetischen Fingerabdruck, der sich wahrscheinlich nicht aus seinen nahen Verwandten entwickelt hat. Wir halten es für sehr wahrscheinlich, dass SARS-CoV-2 als infektiöser Klon entstanden ist, der in vitro zusammengesetzt wurde.“
Der hier vorgestellte Vorschlag sieht vor, dass DEFUSE und das Wuhan-Labor Gain-of-Function-Forschung betreiben, bei der das Genom eines bekannten Virus so verändert wird, dass es eine zusätzliche gewünschte Funktionalität erhält, z. B. eine verbesserte Übertragbarkeit oder Pathogenität (verbessert ist hier das wichtige Wort – es geht nicht darum, eine gleichwertige Pathogenität oder Übertragbarkeit wie bei einem bestehenden Virus zu erreichen, sondern darum, diese neue Funktionalität zu „gewinnen“).
Die der GoF-Forschung (Gain of Function) zugrunde liegende Annahme ist, dass jeder Phänotyp (Eigenschaft oder Funktion) identifiziert und direkt einem oder mehreren spezifischen Genotypen zugeordnet werden kann, die zusammen diese Wirkung in der Zielspezies hervorrufen, und dass durch die Veränderung oder den Ersatz dieser Genotypen auf spezielle Weise ein gewünschter funktioneller Phänotyp geschaffen werden kann. Ein durch die GoF geschaffenes Virus könnte dann als Biowaffe eingesetzt werden oder einen Schritt voraus sein, indem vorausschauende Behandlungen für potenzielle zukünftige Biowaffen oder Pandemieviren geschaffen werden.
Ist die Entwicklung tödlicherer Viren möglich? Der Fluch der Dimensionalität
Ein Gegenargument hierzu wurde vorgebracht von Wu der 1316 Betacoronavirus- und 1378 Alphacoronavirus-Genome analysierte, die vor 2020 gesammelt wurden, und feststellte, dass eine signifikante Teilmenge davon dieselben ungewöhnlichen Endonuklease-Fingerprint-Merkmale (BsmBI/BsaI) aufweist, die mit SARS-CoV-2 in Verbindung gebracht werden. Es könnte also doch nicht so ungewöhnlich sein, und von Bruttel et al. sagte er:
“….die Rückschlüsse können durch die Analyse einer begrenzten Anzahl von Coronavirus-Genomen verzerrt sein und berücksichtigen nicht die Dynamik der Endonuklease-Erkennungsstellen während der viralen Evolution. Hier habe ich eine gründliche Untersuchung der BsmBI/BsaI-Karte in den Genomen von Betacoronavirus, Alphacoronavirus und SARS-CoV-2 durchgeführt.“
„… das Muster der Erkennungsstellen des Restriktionsenzyms BsamBI/BsaI in Alphacoronaviren und Betacoronaviren ist sehr vielfältig, und SARS-CoV-2 ist nicht der einzige Ausreißer in Bezug auf die Anzahl und Position der beiden Endonuklease-Stellen.“
Es sieht also so aus, als ob diese Fingerabdrücke nicht nur für SARS-CoV-2 typisch sind und nicht notwendigerweise mit menschlichen Eingriffen in Verbindung gebracht werden können.
Darüber hinaus untersuchte Wu in einer sehr interessanten Analyse die Möglichkeit einer künstlichen Entwicklung von SARS-CoV-2, wobei er sich auf Gain-of-Function-Insertionen (reverse Genetik) und serielle Passage (das Verfahren, bei dem Bakterien oder ein Virus in Iterationen gezüchtet werden) konzentrierte:
„Die Herausforderung bei der Entwicklung eines neuen Virus liegt nicht in der Methode oder dem Werkzeug für den Genomaufbau, sondern in der Generierung einer umfangreichen Mutantenbibliothek und der Entwicklung eines effizienten Hochdurchsatz-Screening-Systems zur Identifizierung der infektiösesten Klone.
Wu schätzte die Kosten für die Schaffung eines Gain-of-Function-Virus, indem er die Anzahl der Schritte in der natürlichen Evolution von SARS-CoV-2 alpha zur Omicron-Variante verglich und dies auf die Evolution vom Fledermausvirus RaTG135 zu SARS-CoV-2 (unter Verwendung der Anzahl der weltweit mit jeder Variante infizierten Menschen und der entsprechenden Zeiträume)6. Wu beobachtete dies:
„…man kann davon ausgehen, dass es mindestens (80-250 Millionen) experimentelle Iterationen braucht, um ein neues Virus wie SARS-CoV-2 aus RaTG13 zu erzeugen….die Kosten für Laborverbrauchsmaterialien für ein ∼250 Millionen teures Screening- und Testexperiment würden leicht 10 Milliarden Dollar übersteigen…. die Kosten für die Erzeugung eines SARS-CoV-2 würden die volle Investition des Wuhan Institute of Virology für mindestens 100 Jahre erfordern.“
Über die Auswirkungen der Durchführung von Serienpassagen:
„Ein Infektionszyklus in Zellkulturen dauert in der Regel 1 bis 3 Tage, je nach der inokulierten viralen MOI (Multiplicity of Infection). Um gerichtete Mutationen an diesen 1182 Nukleotiden in RaTG13 zu erzeugen, bräuchten wir also mindestens 121 230 Tage (332 Jahre) durch serielle Zellpassage. Aufgrund der längeren viralen Inkubationszeit und des mit der Tierpflege verbundenen Arbeitsaufwands dürfte die serielle Passage bei Tieren eine größere Herausforderung darstellen als bei Zellkulturen. Daher ist es unwahrscheinlich, dass SARS-CoV-2 durch serielles Passaging gewonnen werden kann.
Nach Wus Argumentation wäre es für niemanden möglich, ein neues lebensfähiges Virus zu entwickeln, das eine Reihe von vorbereiteten GoF-Anforderungen erfüllt7. Das DEFUSE-Projekt hätte nicht nur sehr viel Glück haben müssen, um ein Virus zu entdecken, das zuverlässig gezüchtet werden konnte und alle erforderlichen Eigenschaften aufwies, sondern es hätte auch getestet werden müssen, ob es für die Übertragung auf den Menschen geeignet ist und eine erhöhte Pathogenität aufweist.
Ein faszinierender Artikel im Bulletin of Atomic Scientists untersuchte die Gain-of-Function-Forschung und berichtete, dass:
„In der gesamten Biologie ist der genetische Hintergrund wichtig. Dieselbe Mutation kann bei verschiedenen Stämmen zu unterschiedlichen Ergebnissen führen. Das bedeutet, dass die Auswirkungen einiger dieser Mutationen nicht einfach extrapoliert werden können. Dies wiederum deutet darauf hin, dass die Zahl der Mutationen, die ein Virus zu einer pandemischen Bedrohung machen könnten, groß sein könnte, was sowohl für die Gain-of-Function-Forschung als auch für alternative Ansätze ein Problem darstellt. Es gibt eine Vielzahl anderer sehr detaillierter virologische Argumente, warum eine Pandemievorhersage praktisch unmöglich ist.
Die „gain-of-function“-These ist von der Überzeugung geprägt, dass die Virologie mit dem Ingenieurwesen vergleichbar ist; man muss nur die (offensichtlich geringe) Anzahl von Mutationskombinationen definieren, die es einem Virus ermöglichen, zwischen Menschen zu wechseln und sich zu übertragen, und „wir werden es schon herausfinden“. Biologie und Evolution wurden vergessen.
Ron Fouchier, ein Virologe in den Niederlanden und einer der Protagonisten der Gain-of-Function-Influenza, sagte auf einer Konferenz im Jahr 2014, dass er 30 Jahre brauchen könnte, um die Genetik der Übertragung der Vogelgrippe auf den Menschen zu ergründen. Da die meisten von uns nicht weiter als zwei Jahre sehen können, waren 30 Jahre ein Euphemismus für „Ich weiß es nicht“. Die Gain-of-Function-Forschung kann niemals ihr Versprechen einlösen, nämlich die Vorhersage des nächsten pandemischen Grippestamms, um die Entwicklung von präventiven Medikamenten und Impfstoffen zu ermöglichen.“
Wie wir später erörtern werden, haben Experimente zur Erforschung von „wildem“ SARS-CoV-2 die FCS verändert und neue Viren erzeugt. Diese Experimente können durchgeführt werden und wurden auch durchgeführt, aber das bedeutet nicht, dass sie zwangsläufig erfolgreich sind, da niemand weiß, ob die erzeugten Viren selbst lebensfähig sein werden und so hergestellt werden können, dass sie die funktionelle Anforderung „übertragbar und tödlich“ (oder eine andere) erfüllen. Wenn die Anforderungen an ein für den Menschen pathogenes Virus gestellt werden, sind letztlich8 dann sind Challenge-Studien erforderlich, nicht an Mäusen, sondern an Menschen. Soweit wir wissen, wurden solche Studien nicht durchgeführt, und auf jeden Fall sind alle Challenge-Studien, die mit wildem SARS-CoV-2 durchgeführt wurden, fehlgeschlagen (hier), und zwar ziemlich kläglich. Daher können alle Behauptungen über die Funktionsfähigkeit teilweise in vitro oder in Tiermodellen bestätigt werden, aber nicht vollständig beim Menschen oder in der Gesellschaft insgesamt. Alles, was wir wissen, ist, dass Viren produziert werden können, aber man kann nicht garantieren, dass sie „viral“ werden oder „tödlich“ sind9. Wus Analyse legt nahe, dass das ganze Unterfangen zum Scheitern verurteilt, wenn nicht sogar unmöglich ist.
Gibt es eigentlich irgendeinen Beweis dafür, dass jemals ein lebensfähiges Virus entwickelt wurde, das einer vorbereiteten Funktionsspezifikation entspricht und eine Pandemie auslösen könnte? Soweit uns bekannt ist, gibt es keine eindeutigen Erfolge. Masters und Perlman führen zum Beispiel einen Fall von Feline Enteric Coronavirus (FeCoV) an, bei dem mit Hilfe der reversen Genetik S-Proteine von (vermutlich) virulenten und avirulenten Stämmen ausgetauscht wurden, aber die Auswirkung auf die Pathogenese ist zugegebenermaßen unbekannt, da die Experimente nur in vitro durchgeführt wurden.
Eine frühere Kontroverse im Jahr 2014 drehte sich um die GoF-Forschung im Zusammenhang mit drei Experimenten zur erzwungenen Evolution, die an Frettchen durchgeführt wurden, die mit H5N1- und H7N1-Vogelgrippeviren infiziert waren (dokumentiert hier, hier und hier). Die ersten beiden GoF-Experimente an Frettchen haben gezeigt, dass die Übertragbarkeit verbessert werden könnte (aber möglicherweise nicht für den Menschen vorhersagbar ist), allerdings erwiesen sich die erzeugten GoF-Varianten als nicht ausreichend replikationsfähig.
In der dritten der oben zitierten Studien (von Sutton et al.) infizierten die Forscher vier Frettchen direkt, führten serielle Passageexperimente an diesen durch und versuchten dann, naive Frettchen zu infizieren. Sie berichteten, dass ein hoch pathogenes Vogelgrippevirus des Subtyps H7N1, das sich in der Vergangenheit nicht an Säugetiere angepasst hat, im Frettchenmodell des Influenzavirus über die Luft übertragen werden kann. Dies sieht alarmierend aus, und man könnte viel daraus machen, dass alle vier direkt infizierten Frettchen eingeschläfert wurden, weil sie Anzeichen einer schweren Krankheit aufwiesen. Diese Frettchen wurden jedoch nicht wie üblich ein einziges Mal, sondern mehrere Male in Serie infiziert. Eine alternative Erklärung für die Euthanasie dieser Tiere ist daher nicht unbedingt eine erhöhte Pathogenität, sondern wahrscheinlich, dass ihr Immunsystem durch wiederholte Reinfektionen einfach überfordert war. Die Autoren berichteten über eine erfolgreiche Anpassung der Vogelgrippe, so dass sie in Frettchen über die Luft übertragen werden konnte, kamen jedoch zu dem Schluss, dass das erzeugte GoF-Virus möglicherweise nicht die für eine Pandemie erforderliche Übertragungseffizienz erreicht hat. Angesichts der geringen Stichprobengröße war die Frage nach einer erhöhten Pathogenität bei Frettchen, die mit der GoF-Variante infiziert waren, nicht eindeutig zu beantworten10 Die Gesamtheit der Beweise deutet jedoch darauf hin, dass das GoF-Virus in den Lungen und Bronchien usw. weniger pathogen war als das Wildtyp-Virus (wie zu erwarten).
In Anbetracht der Tatsache, dass Loss-of-Function (LoF)-Experimente, bei denen Merkmale eines Virus entfernt werden, gang und gäbe sind und der Entwicklung von Impfstoffen mit abgeschwächten Viren gleichkommen, ist diese Studie von Fauci selbst ein weiterer Sargnagel im herrschenden Narrativ. Er räumt ein, dass die Entwicklung von Impfstoffen für Atemwegsviren allein aufgrund der schnellen Virusreplikation, der Fehlerquote bei der Mutation und des Antigendrift schwierig ist. Er sagt dies (nach der Einführung und Injektion der Covid-19-Impfstoffe in Milliarden von Menschen!):
“Dauerhaft schützende Impfstoffe gegen nicht-systemische mukosale Atemwegsviren mit hoher Sterblichkeitsrate haben sich bisher den Bemühungen um die Entwicklung von Impfstoffen entzogen.”
Wenn diese Merkmale die Entwicklung von Impfstoffen für Atemwegsviren schwierig, wenn nicht gar unmöglich machen, dann machen sie auch die Virusentwicklung mittels Gain-of-Function zu einer ebenso großen, wenn nicht sogar noch größeren Herausforderung (vielleicht sogar genauso unmöglich).
In der Mathematik nennt man dies den Fluch der Dimensionalität. Bei vielen Problemen, bei denen jede Variable (Proteine, Genome, Zellen, Virionen) mehrere Werte annehmen kann, muss eine riesige Anzahl von Wertekombinationen berücksichtigt werden, was zu einer kombinatorischen Explosion führt. Dies führt zu einer exponentiellen Menge von Möglichkeiten, von denen jede einzelne berücksichtigt und bewertet werden muss. Im biologischen Bereich sind die Dinge sogar noch schwieriger, weil es keine Monotonie gibt – jeder Phänotyp (Eigenschaft oder Funktion) kann nicht direkt einem einzigen spezifischen Genotyp zugeordnet werden; daher ist das Problem nicht reduzierbar, und es gibt keinen Algorithmus, der den Raum der Möglichkeiten durchsuchen und die einzige Genotypsequenz identifizieren kann, die den gewünschten Phänotyp liefert. RNA-Viren sind Mutationsmaschinen mit dem Potenzial, sich an einen neuen Wirt anzupassen. Selbst wenn eine stabile Virusfunktion isoliert wird, wird sie schnell mutieren und diese neu gewonnene Funktionalität verlieren (Wain-Hobson).
Die Schaffung eines wirklich pandemieauslösenden Coronavirus würde daher wahrscheinlich das Re-Engineering eines oder mehrerer Coronaviren erfordern und wäre mit der GoF-Forschung einfach nicht möglich. Nur die Zeitskala der natürlichen Evolution könnte die Zeit und den Raum bieten, die erforderlich sind, um sowohl lebensfähige als auch nicht lebensfähige Kombinationen in einer Weise zu schaffen, die der Mensch nicht leisten kann. Die Evolution (oder Gott) ist notwendig, um den Fluch der Dimensionalität zu besiegen.
Experimente zum Gewinn (oder Verlust) von Funktionen
Bei der Erforschung von SARS-CoV-2 kommt natürlich das Handwerkszeug zum Einsatz, einschließlich der Anwendung der reversen Genetik, um Variationen des Virus zu erzeugen. Solche Variationen könnten Änderungen an der Furin-Spaltstelle und dem Spike-Protein umfassen11.
Im Januar 2021 Johnson et al. ein LoF-Experiment mit Hilfe der reversen Genetik durch und beobachteten, dass vier mit dem Wildvirus infizierte Hamster im Vergleich zum LoF-Arm an Gewicht verloren und einige Anzeichen einer Infektion zeigten. Außerdem stellten sie fest, dass das LoF-Virus entgegen den Erwartungen effizienter repliziert wurde. Als sie das Experiment mit transgenen Mäusen wiederholten, fanden sie keine Unterschiede in der Viruslast, aber aufgrund der Gesamtheit der Beweise kamen sie zu dem Schluss, dass die LoF-Mutante im Vergleich zum Wildtyp-Virus eine geringere Krankheit und Replikation zu einem frühen Zeitpunkt nach der Infektion verursachte. Sie verwendeten auch transgene Mäuse mit hACE2-Rezeptoren (12 pro Arm), um die Pathogenität zu untersuchen, und stellten fest, dass das Wildvirus zwar einen stärkeren Gewichtsverlust verursachte, aber keine Unterschiede in der Viruslast in der Lunge oder im Gehirn aufwies. Um die funktionellen Auswirkungen auf die Lunge zu bestimmen, wurden die Mäuse mechanisch beatmet, um biophysikalische Parameter zu messen, wobei die Wildtyp-Mäuse mehr Schäden aufwiesen. Die Histopathologie bestätigte dies.
Im Juli 2021 Davidson et al. klonierten WT (Wildtyp) SARS-CoV-2 und erzeugten Varianten mit GoF-Verbesserungen durch Engineering von Spike-Varianten. Die Laborergebnisse zeigten, dass LoF, insbesondere die Deletion der Furin-Spaltstelle, zu höheren Infektionsraten menschlicher Endothelzellen (Atemwege) führte, und als sie die Virusübertragung an acht Frettchen testeten, stellten sie fest, dass zwei der vier Frettchen, die mit absichtlich mit WT SARS-CoV-2 vorinfizierten Frettchen zusammenlebten, Anzeichen einer Infektion zeigten. Und 0 der 4 Frettchen, die mit absichtlich mit der GoF-Variante vorinfizierten Frettchen zusammen gehalten wurden, zeigten Anzeichen einer Infektion. Sie sagen:
„Wir zeigen, dass sich ein Virus mit deletiertem Furin-CS im Gegensatz zu WT-SARS-CoV-2 nicht mit hohen Titern in den oberen Atemwegen von Frettchen repliziert und nicht auf Sentinel-Tiere übertragen wird, was mit ähnlichen Experimenten mit Hamstern übereinstimmt.”
Diese Studie basiert auf einer so kleinen Stichprobe, dass die Ergebnisse nicht signifikant sind, und wir wissen nicht, ob die Infektion einen Einfluss auf die Pathogenität hatte. Keines der Frettchen aus beiden Versuchen zeigte nennenswertes Fieber oder Gewichtsverlust. Bei der Verwendung einer „konkurrierenden“ Mischung aus 70 % WT und 30 % LoF-Mutanten waren die Ergebnisse überhaupt nicht eindeutig. Bei einigen Frettchen dominierte die LoF-Mutante in den mit der Mischung geimpften Frettchen, und von den vier gemeinsam untergebrachten Frettchen wurde nur eines durch Übertragung infiziert. Das macht die Ergebnisse schwer zu interpretieren.
In einer ähnlichen Studie im Februar 2021 verwendeten Zhu et al. naive Hamster, die zusammen mit einem einzigen infizierten Hamster untergebracht waren, und untersuchten die Veränderungen an der Furin-Spaltstelle anhand von sechs Hamstern in jedem Arm (Wildtyp und LoF-Arm). Trotz der heroischen Anwendung von statistischem Geschick bei einer so kleinen Stichprobengröße sind die Ergebnisse – nicht überraschend – schlecht und nicht überzeugend. Die mit dem Wildtyp-Virus infizierten Hamster verloren etwa 10 % an Gewicht, während die mit der LoF-Variante infizierten Hamster an Gewicht zunahmen; es scheint keinen Grund zu geben, warum das Gewicht zunehmen sollte, was auf eine experimentelle Störung hindeutet. Die anderen Ergebnisse waren nicht überzeugender, was angesichts der geringen Stichprobengröße nicht überrascht.
Im Jahr 2024 Valleriani et al. eine viel größere Studie mit 150 transgenen Mäusen durch, die den hACE212 Rezeptor exprimieren, unter der Annahme, dass diese für eine SARS-CoV-2-Infektion empfänglich sind und in der Lage sind, eine schwere Krankheit zu entwickeln (daher sind sie anfällig für Spikes). Auch hier wurde der Vergleich mit einem LoF-Arm durchgeführt. Die LoF-infizierten Mäuse wiesen eine geringere Ausscheidung, eine geringere Virulenz auf Lungenebene und mildere Lungenläsionen auf. Die Mäuse im LoF-Arm hatten eine etwas höhere Überlebensrate und niedrigere klinische Werte als der Wildtyp des Virus.
Nimmt man diese experimentellen Ergebnisse für bare Münze, so deuten sie darauf hin, dass die LoF-Variante eine höhere Übertragbarkeit, aber eine geringere Pathogenität aufweist, während die GoF-Variante eine geringere Übertragbarkeit und eine höhere Pathogenität aufweist13. Dies verdeutlicht den inhärenten Kompromiss zwischen diesen funktionellen Merkmalen: Man kann nicht gleichzeitig ein hoch übertragbares und ein hoch tödliches Virus haben. Dies könnte bestätigen, wie schwierig es ist, den Fluch der Dimensionalität zu überwinden – es ist nahezu unmöglich, Änderungen vorzunehmen, die beide Funktionen erfüllen können.
Beachten Sie, dass bei all diesen Experimenten keine Kontrollen mit anderen Coronaviren oder Influenzaviren zum Vergleich mit SARS-CoV-2 durchgeführt wurden. Wir wissen einfach nicht, wie diese LoF- oder Wildvirus-Ergebnisse mit gewöhnlichen Erkältungs- oder Influenzaviren zu vergleichen sind. Wir wissen auch nicht, was passieren würde, wenn wir FCS oder andere Gene aus diesen anderen Coronaviren entfernen würden. Würden sie pathogener oder übertragbarer werden? Sind sie bereits pathogener oder weniger pathogen? Außerdem wurden keine Tests durchgeführt, um bakterielle Lungenentzündungen oder andere konkurrierende Krankheitserreger auszuschließen, was die Experimente beeinträchtigt haben könnte.
Bei vielen anderen Coronaviren wurde die Pathogenese im Zusammenhang mit Spike bereits diskutiert, z. B. in der Arbeit von Millet und Whitaker. Die Korrelation zwischen der Pathogenese und den Spikes bestimmter Coronaviren ist jedoch verzerrt, einfach weil sie auf stark verwirrten Beobachtungsdaten beruht (z. B. wird der Spike für MERS-CoV als gefährlicher angesehen, weil er mit einer „Pandemie“ in Verbindung gebracht wird, während die Spikes von Coronaviren, die nicht damit in Verbindung gebracht werden, dies nicht tun). Dies ist ein wichtiger Punkt – wenn die Mortalitätsdaten früherer Pandemien durch Behandlungsprotokolle und medizinische Nachlässigkeit verzerrt sind, woher wissen wir dann, dass der Spike oder ein anderes Merkmal wirklich der Grund für die Sterblichkeit ist?
Die großen Unterschiede bei den Symptomen von SARS-CoV-2, die in verschiedenen Ländern bei Patienten gemeldet wurden, die angeblich mit demselben Virus infiziert waren, sollten die Alarmglocken schrillen lassen, da dies darauf hindeutet, dass die gemeldeten Symptome durch den Aufenthaltsort und die Testergebnisse verfälscht wurden (Neil et al.). Es ist eine Umkehrung der Realität, wenn behauptet wird, dass dasselbe Virus unterschiedliche Symptome verursacht, die an verschiedenen geografischen Orten auftreten, während in der Vergangenheit unterschiedliche Viren (insbesondere solche, die mit Atemwegserkrankungen in Verbindung gebracht werden) weltweit dieselben Symptome verursacht haben.
Veröffentlichte GoF- und LoF-Experimente sind nicht überzeugend in Bezug auf unsere Fähigkeit, Viren so zu manipulieren, dass sie funktionale Anforderungen erfüllen. Das Fehlen von Vergleichskontrollen reicht aus, um dies zu bestätigen, ebenso wie Probleme im Zusammenhang mit der Heterogenität von Symptomen bei einem vermeintlich manipulierten Virus. Es bleibt daher eine offene Frage, ob alle Coronavirus-Infektionen für den Menschen relativ unpathogen sind, mit oder ohne Spike oder FCS oder andere Einschübe, ob künstlich oder natürlich, und dass SARS-CoV-2 in dieser Hinsicht nicht anders sein könnte.
Widersprüchliche Beweise für den Ursprung von Zoonosen
Wenden wir uns den tierischen Reservoiren zu, um Schätzungen über die phylogenetischen Ursprünge des Virus zu untersuchen. Munnik et al. berichten, dass SARS-CoV-2 Hauslöwen, Tiger, Katzen, Hunde, Frettchen, Hamster, Spitzhörnchen, Kaninchen und sogar Tiger in Zoos infizieren kann. Aber keine Schweine oder Geflügel (bis jetzt!)!
Sie untersuchten einen zoonotischen Ausbruch der Infektion in niederländischen Nerzfarmen im Jahr 2020.
„Bei den Sequenzen aus einigen Nerzfarmen wurde eine hohe Diversität festgestellt, was wahrscheinlich auf mehrere Generationen von Virusinfektionen bei Tieren zurückzuführen ist, bevor der Anstieg der Sterblichkeit festgestellt wurde. ….
… hat die Untersuchung keine gemeinsamen Faktoren ermittelt, die die Ausbreitung von Betrieb zu Betrieb erklären könnten.“
“Das könnte bedeuten, dass das Virus bereits einige Zeit vor seiner Entdeckung in Nerzfarmen zirkulierte.“
Könnte es sein, dass das Virus bereits in dem Nerz war?
Im Jahr 2020 Boni et al. die evolutionären Ursprünge von SARS-CoV-2 untersucht und berichtet:
„SARS-CoV-2 selbst ist keine Rekombination eines der bisher entdeckten Sarbecoviren, und sein rezeptorbindendes Motiv, das für die Spezifität für menschliche ACE2-Rezeptoren wichtig ist, scheint ein uraltes Merkmal zu sein, das mit Fledermausviren geteilt wird und nicht erst kürzlich durch Rekombination erworben wurde. ….
Das Datum der Divergenz zwischen SARS-CoV-2 und dem Fledermaus-Sarbecovirus-Reservoir wurde auf 1948 (95% höchste posteriore Dichte (HPD): 1879-1999), 1969 (95% HPD: 1930-2000) und 1982 (95% HPD: 1948-2009), was darauf hindeutet, dass die Linie, die SARS-CoV-2 hervorgebracht hat, seit Jahrzehnten unbemerkt in Fledermäusen zirkuliert.“
Wenn es seit Jahrzehnten unbemerkt in Fledermäusen zirkuliert, warum sollte es dann nicht auch schon seit Jahrzehnten in Menschen zirkulieren? Und mit Jahrzehnten meinen wir irgendeinen Zeitpunkt zwischen 1879 und heute?
Dieses faszinierende Ergebnis wurde bisher weitgehend ignoriert. In seiner viel zitierten Arbeit aus dem Jahr 2024 über die Entstehung und Entwicklung von SARS-CoV-2 erwähnt Holmes diese Studie kaum und zitiert auch ihre Schlussfolgerungen nicht.
Markov et al. untersuchten auch die Entwicklung der SARS-CoV-2-Prüfung:
„…die selektiven Kräfte, die wahrscheinlich die Entwicklung einer höheren Übertragbarkeit und in einigen Fällen eines höheren Schweregrades während des ersten Jahres der Pandemie angetrieben haben, und die Rolle der Antigenentwicklung während des zweiten und dritten Jahres.“
Sie fanden:
„Nach dem Auftreten von SARS-CoV-2 beim Menschen schien das Virus in den ersten fast acht Monaten nur eine begrenzte scheinbare Evolution zu zeigen. Dies war zum Teil auf die relativ kleine globale Viruspopulation zurückzuführen, während die Ausbreitung noch nicht allgegenwärtig war, und später auf nicht-pharmazeutische Interventionen in vielen Teilen der Welt, und zum Teil ein Artefakt der Unterbeprobung des Virus.“
Es dauerte acht Monate, bis die ersten abweichenden SARS-CoV-2-Linien auftauchten…., was aus evolutionärer Sicht einen Wendepunkt in der Pandemie darstellt. Die ersten drei dieser Linien, die später als VOCs Alpha, Beta und Gamma bezeichnet wurden, entstanden unabhängig voneinander in verschiedenen Teilen der Welt und waren das Ergebnis rätselhafter höherer Evolutionsraten. Die schiere Anzahl der Mutationen, die bei den VOCs auftraten, ist aus evolutionärer Sicht besonders auffällig.“
Dies deutet darauf hin, dass das Virus zu Beginn der Pandemie eine in jeder Hinsicht identische, standardisierte Kopie war, aber Ende 2020 gab es viele davon. In den Nerzfarmen in den Niederlanden finden wir jedoch Nerze mit einer sehr hohen Diversität der beprobten Sequenzen, was darauf hindeutet, dass das Virus schon lange vor 2020 im Tierreservoir vorhanden war. Wie kann die menschliche Bevölkerung mit einer standardisierten Version von SARS-CoV-2 infiziert sein, während bei Tieren Varianten gefunden werden und bei Fledermäusen überhaupt keine SARS-CoV-2-Variante vorkommt? Und eine sorgfältige phylogenetische Analyse deutet darauf hin, dass Vorläufer des Virus schon seit Jahrzehnten (vielleicht seit 1879?) unbemerkt in Fledermäusen zirkulieren.
Despres et al. untersuchten die Wildtiere in Vermont, USA, um festzustellen, ob sie SARS-CoV-2 nachweisen konnten, und fanden bei den Wildtieren im gesamten Bundesstaat, einschließlich Rehen, kein einziges Exemplar, obwohl in den meisten veröffentlichten nordamerikanischen Studien SARS-CoV-2 in den dortigen Rehpopulationen nachgewiesen wurde. Als Grund dafür nannten sie „umweltbedingte und anthropogene Faktoren“.14.
Kumar et al. führten eine globale Sequenzierung von SARS-CoV-2 in Kombination mit Berechnungsmethoden durch, um den jüngsten gemeinsamen Vorfahren des Virus zu ermitteln, was darauf hindeutet, dass der Patient Null in Wuhan nicht der Indexfall war und auch nicht die Ursache für alle menschlichen Infektionen. Daraus lässt sich schließen, dass der Vorläufer des Virus sich bereits Monate vor dem Ausbruch in Wuhan weltweit verbreitet hat. Sie behaupteten, das Vorläufergenom (Root-Genom) von SARS-CoV-2 durch Messung der „Coronavirus-Diversität“ identifiziert zu haben.
Eine kritische Einschränkung der phylogenetischen Analyse ist die Annahme, dass es keine Rekombination zwischen Virusarten und -unterarten gibt. Stattdessen wird davon ausgegangen, dass die Variation „innerhalb“ und nicht „zwischen“ den Arten stattfindet und dass genetische Sequenzen, die in einem oder mehreren Viren vorkommen, einen gemeinsamen Vorfahren in einem anderen haben könnten. In Anbetracht dessen hat das Konzept einer „Wurzel“ des Stammbaums keine Gültigkeit. Auch die Annahme, dass es eine vorhersagbare molekulare Uhr gibt, die aus ähnlichen Coronaviren abgeleitet werden kann und die selbst im Laufe der Zeit konstant bleibt, wird durch diese Aspekte ernsthaft in Frage gestellt. Das ICTV gibt dies sogar zu.
In Anbetracht dessen ist es vielleicht optimistisch zu glauben, dass die Entdeckung von Antikörpern gegen SARS-CoV-2 in Abwasser und anderen Proben aus dem Jahr 2019 (schön zusammengefasst hier), in irgendeiner Weise endgültig sein könnte. Das liegt daran, dass Antikörper Epitope abdecken, die von verschiedenen Viren gemeinsam genutzt werden, so dass unser Immunsystem in der Lage ist, einen äußerst vielfältigen Angriff zu starten, um verschiedene Viren und Varianten zu zerstören, ohne sich selbst zu erschöpfen (wie sonst könnte es die kombinatorische Komplexität eines sich ständig weiterentwickelnden Virenschwarms besiegen?) Es ist daher möglich, wenn auch nicht sicher, dass die angeblich spezifischen Antikörper gegen SARS-CoV-2 nicht gegen SARS-CoV-2 selbst, sondern gegen einen Vorläufer von SARS-CoV-2 entwickelt wurden, der die gleichen Epitope enthält, oder gegen andere ähnliche Coronaviren15.
Hier gibt es zu viele Widersprüche. Weder der phylogenetischen Analyse noch den Antikörperuntersuchungen kann man in vollem Umfang vertrauen, wenn es um die endgültige Herkunft geht, und es gibt eine vielbeachtete Schätzung der evolutionären Ursprünge von SARS-CoV-2, die bis ins Jahr 1879 zurückreicht. Die Möglichkeit, dass der Erreger bereits seit einiger Zeit in der Tierpopulation endemisch ist, ist daher nicht von der Hand zu weisen, denn er ist das Ergebnis einer Koevolution von Viren in einem Schwarm.
Die Hypothese der zoonotischen Endemie ist also nicht falsifiziert worden (und um fair zu sein, kann sie es wahrscheinlich auch nicht sein, weil die Grundlagen so schlecht verstanden sind).
GoF-Forschung ist Routine und beinhaltet die Erstellung von (infektiösen?) Klonen
Interessanterweise befassen sich Millet und Whitaker in ihrem (von den US-amerikanischen NIH finanzierten) Papier aus dem Jahr 2014 mit dem Potenzial der FCS-Forschung und plädieren eindeutig für eine darauf ausgerichtete GoF-Forschung, was zeigt, dass dies ein gemeinsames Forschungsthema in Coronavirus-Forschungskreisen ist. Sie sagen es ausdrücklich:
„Insgesamt scheint es wahrscheinlich, dass die Modulation einer der beiden Protease-Spaltstellen durch Coronaviren einen tiefgreifenden Einfluss auf den Krankheitsverlauf haben kann, je nach Coronavirus.”
Der FCS ist ein Beispiel für eine Proteaseschnittstelle. Dies deutet darauf hin, dass die auf die FCS konzentrierte GoF/LoF-Forschung keine Besonderheit des DEFUSE-Projekts ist, was die Hypothese, dass die Eigenschaften des „neuartigen“ Virus aufgrund der Tatsache, dass im Rahmen des DEFUSE-Projekts genau in diesem Bereich geforscht wurde, bedeuten müssen, dass es von Menschenhand geschaffen wurde, erheblich entkräftet.
Bruttel et al. sagt dies über die routinemäßige Anwendung der reversen Genetik zur Erzeugung von Mutationen (d. h. zur Veränderung der Funktion):
„Die Herstellung eines reversen genetischen Systems aus einem Wildtyp-CoV erfordert die Zerlegung des 30 kb großen coronaviralen Genoms in 5-8 Fragmente, von denen jedes typischerweise kürzer als 8kb ist (Almazán et al. 2006; Becker et al. 2008; Scobey et al. 2013; Zeng et al. 2016; Cockrell et al. 2017; Hu et al. 2017). Um ein reverses genetisches System zu entwerfen, modifizieren Forscher häufig ihre synthetischen DNA-Konstrukte aus den Wildtyp-Virusgenomen, indem sie synonyme Mutationen einführen, die die Erkennungsstellen der Restriktionsenzyme verändern, ohne die Fitness der resultierenden infektiösen Klone wesentlich zu beeinträchtigen.“
Die erste Studie von Almazán et al. beschreibt die Herstellung eines cDNA-Klons (komplementäre DNA) in voller Länge aus SARS. Ein cDNA-Klon enthält die gesamte genetische Information des Virus, die für die Replikation, Transkription und Translation erforderlich ist. Anders ausgedrückt, sie haben das Potenzial, infektiös zu sein, genau wie das ursprüngliche Virus, das sie geklont haben. Sie benutzen es:
„…..für die Wiederherstellung infektiöser Viren und es wurde für die Erzeugung einer großen Sammlung von Deletionsmutanten von SARS-CoV verwendet.
Eine Deletionsmutante ist eine genetische Anomalie, bei der ein Segment eines Chromosoms oder einer DNA-Sequenz während der DNA-Replikation ausgelassen wird, was zum Fehlen bestimmter Nukleotide oder ganzer Chromosomensegmente führt. Dies kann zu einer veränderten Genfunktion oder -expression führen. Dies ist die GoF.
Schauen wir uns an Becker et al. (Ralph Baric ist Mitverfasser dieses Papiers von 2008):
„Die Bestimmung der voraussichtlichen Wege, auf denen sich Zoonosen entwickeln und zu Krankheitserregern für den Menschen werden, ist entscheidend für die Vorhersage und Kontrolle sowohl natürlicher als auch absichtlicher Pandemien. Die Vorhersage vertretbarer Übertragungswege vom Tier auf den Menschen wird jedoch durch die Schwierigkeiten bei der Identifizierung von Reservoir-Spezies, der Kultivierung zoonotischer Organismen in Kulturen und der Isolierung von Genomen in voller Länge für das Klonen und genetische Studien behindert. Die Möglichkeit, aus synthetisch hergestellten cDNAs rekonstituierte Erreger zu entwickeln und zu gewinnen, hat das Potenzial, diese Hindernisse zu überwinden, indem sie Studien zur Replikation und Pathogenese ohne Identifizierung von Reservoir-Spezies oder Kultivierung von Primärisolaten ermöglicht. Hier berichten wir über das Design, die Synthese und die Wiederherstellung der größten synthetischen replizierenden Lebensform, eines 29,7 KB großen Fledermaus-Coronavirus, das dem schweren akuten respiratorischen Syndrom (SARS) ähnelt (Bat-SCoV), einem wahrscheinlichen Vorläufer der SARS-CoV-Epidemie. Um einen möglichen Entstehungsweg vom nicht kultivierbaren Bat-SCoV zum menschlichen SARS-CoV zu testen, haben wir ein Konsensus-Bat-SCoV-Genom entworfen und die Spike-Rezeptor-Bindungsdomäne (RBD) von Bat-SCoV durch die RBD von SARS-CoV (Bat-SRBD) ersetzt. Bat-SRBD war in Zellkulturen und in Mäusen infektiös und wurde durch Antikörper, die sowohl für Fledermaus- als auch für menschliche CoV-Spike-Proteine spezifisch sind, effizient neutralisiert. Die rationelle Entwicklung, Synthese und Gewinnung hypothetischer rekombinanter Viren kann zur Untersuchung der Mechanismen der artenübergreifenden Übertragung von Zoonosen genutzt werden und hat ein großes Potenzial zur Unterstützung einer schnellen Reaktion der öffentlichen Gesundheit auf bekannte oder vorhergesagte neue mikrobielle Bedrohungen.“
Dabei handelt es sich eindeutig um GoF-Forschung, die 12 Jahre vor der SARS-CoV-2-Pandemie veröffentlicht wurde.
Hier ist der Titel der Arbeit von Cockrell et al. (2018):
„Ein mit Spikes modifizierter infektiöser Klon des Coronavirus des Mittleren Ostens (MERS-CoV) löst bei infizierten Rhesusaffen eine leichte Atemwegserkrankung aus“.
Dies ist GoF-Forschung.
Ebenso ist zu beachten, dass sich die Studien zu LoF, die wir bereits zuvor erörtert haben, im Prinzip nicht von GoF unterscheiden. Die GoF-Forschung ist also noch nicht abgeschlossen.
Es sollte auch klar sein, dass das Klonen von Viren in der virologischen Forschung und Entwicklung einfach und sogar ziemlich routinemäßig stattfindet. So heißt es zum Beispiel in der Studie von Bruttel:
„Die BsaI/BsmBI-Karte von SARS-CoV-2 ist anomal für ein wildes Coronavirus und stammt wahrscheinlich von einem infektiösen Klon, der als effizientes Reverse-Genetik-System entwickelt wurde. Die Forschungsziele und die Laborlogistik der infektiösen Klontechnologie können in den Genomen infektiöser Klone einen bisher unbekannten Fingerabdruck hinterlassen“.
Masters und Perlman diskutieren ausführlich und unumstritten über Klone.
Ein von der Obama-Regierung angekündigtes Moratorium wurde 2017 nach drei Jahren (2014 – 2017) aufgehoben, und trotz des Moratoriums hat die US-Regierung weiterhin einige GoF-Experimente bewertet und finanziert (hier). Es handelt sich also keineswegs um ein Verbot.
Es scheint also so zu sein, dass die GoF-Forschung, wie der DEFUSE-Vorschlag, Routine war und immer noch ist. Es sieht in der Tat so aus, als ob dies weltweit in vielen Forschungsgruppen, die sich mit Coronaviren befassen, Standardpraxis ist.
Wenn nicht durch ein Leck im Labor oder auf dem Markt, wie ist „es“ dann entstanden?
Zu den tierischen Ursprüngen von SARS-CoV-2 äußerte sich Wu wie folgt:
„Kürzlich entdeckten Wang et al. eine hohe Häufigkeit von mit Säugetieren assoziierten viralen Koinfektionen und identifizierten 12 Viren, die von verschiedenen Fledermausarten gemeinsam genutzt werden, durch eine Meta-Transkriptomanalyse von 149 individuellen Fledermausproben in Yunnan, China. Die Autoren fanden auch zwei Coronaviren, die eng mit SARS-CoV-2 verwandt sind (92%-93% genetische Identität), wobei sich nur fünf Aminosäuren in der rezeptorbindenden Domäne des einen Genoms vom Wuhan-Hu-1-Stamm unterscheiden. Diese Ergebnisse deuten darauf hin, dass virale Koinfektionen und Spillover bei Fledermäusen weit verbreitet sind, was die hohe Zahl rekombinanter Ereignisse bei Coronaviren erklärt und darauf hindeutet, dass SARS-CoV-2 durch rekombinanten Austausch zwischen mehreren verwandten Genomen entstanden ist.“
Nun sollten wir offenlegen, dass Wu 2012 seinen Abschluss am Wuhan Institute of Virology gemacht hat und er daher als mit einem Interessenkonflikt behaftet werden könnte, aber seine Analyse scheint sich nicht auf neue Forschungsergebnisse zu stützen und extrapoliert einfach das, was derzeit als aktueller Stand der Praxis in der Virologie gilt. Außerdem ist er derzeit an der Universität von Texas tätig.
Natürlich kann keine der beiden Seiten sagen, wie es dazu kam, dass SARS-CoV-2 beim Menschen entdeckt wurde, aber es ist nicht ausgeschlossen, dass es weder vor kurzem in der Natur entstanden ist noch aus einem Labor stammt, sondern dass es schon seit geraumer Zeit in der menschlichen Bevölkerung oder in der Natur herumgeistert, bis es entdeckt wurde.
Betrachten wir die Erklärung für das einzelne Zoonoseereignis auf dem Huanan-Meeresfrüchtemarkt in Wuhan irgendwann zwischen Oktober und Dezember 2019 (molekulare Epidemiologie beschrieben hier). Erscheint es unwahrscheinlich, dass das Virus spontan auf dem Nassmarkt auftauchte, während gleichzeitig die GoF-Forschung in Wuhan lief? Wie groß ist die Wahrscheinlichkeit, dass die Natur genau an diesem Ort und zu dieser Zeit ein neues Virus erschaffen hat? Die Wahrscheinlichkeit wäre astronomisch gering, vor allem, wenn man bedenkt, dass es Fledermäuse, Menschen und Schuppentiere betreffen könnte.
Das lückenhafte, punktuelle Ausbreitungsmuster von SARS-CoV-2 spricht für alternative Erklärungen für den Ursprung von SARS-CoV-2: Die Sterblichkeitsrate stieg im Frühjahr 2020 erst an, nachdem Abriegelungsmaßnahmen eingeführt wurden, und nicht vorher, und die Morbidität wurde erst mit der Einführung von PCR-Tests als einem neuen Virus zuzuschreiben erkannt. (Rancourt, Engler, Pospichal).
Wir können daher über alternative Theorien spekulieren, von denen zwei die bereits existierende Endemizität und die absichtlich hergestellten Virusklone sind, um den Eindruck einer Pandemie zu erwecken. Die erste Theorie besagt, dass die Endemie in der ganzen Welt bereits vorhanden ist (unterstützt durch die bestehende T-Zellen-Immunität und andere Faktoren). Die zweite Theorie besagt, dass der Erreger in einem Labor in Wuhan hergestellt wurde, wobei es sich nicht notwendigerweise um ein Leck in einem Labor in Wuhan handeln muss, sondern dass „etwas“ absichtlich zuerst in Wuhan und dann an mehreren Orten weltweit freigesetzt wurde. Diese Theorien müssen sich nicht gegenseitig ausschließen, aber um sie zu verwirklichen, müssten sie mit einem Projekt verbunden sein, das ein hohes Maß an Verlogenheit und Erfindungsreichtum erfordert.
Vorbestehende Endemie:
- Stellen Sie sich vor, Sie kommen der Wissenschaft zuvor und entdecken ein Coronavirus, das beim Menschen bereits endemisch ist, aber nicht bei Fledermäusen oder Kamelen (vielleicht hat es sich in ferner Vergangenheit zoonotisch ausgebreitet). Sie halten dies geheim, weil Sie wissen, dass dies eine günstige Gelegenheit ist, wenn Sie erfolgreich einen Köder auslegen können. Sie beantragen GoF-Forschungsgelder für die Entwicklung neuer Viren zum Nutzen der Menschheit (Impfstoffe usw.). Sie verwenden das neu entdeckte Virus in Ihrer Forschung und spielen vielleicht sogar mit einigen kleinen Veränderungen herum. Sie sind zuversichtlich, dass Sie diese in Ihrem Antrag auf Fördermittel angeben können, da sie sich später als nützlich erweisen könnten. Sie wissen jedoch, dass GoF unglaublich schwierig ist (was Sie aber weder Ihren Geldgebern noch der Öffentlichkeit gegenüber zugeben), und Sie können es daher nicht tödlicher oder übertragbarer machen, als es derzeit ist (was ohnehin nicht viel ist, da es vielleicht schon seit Äonen im Hintergrund ist). Als Nächstes wird ein Impfstoff für dieses Virus entwickelt (der ebenfalls nicht funktioniert, aber das ist eine andere Geschichte). Es bliebe dann nur noch, einen Test für dieses Virus zu entwickeln, damit es von der übrigen Menschheit entdeckt werden kann, so dass der Eindruck entsteht, es sei plötzlich aufgetaucht, und angesichts seiner Endemizität eine Pandemie vorzutäuschen, um den Verkauf von Impfstoffen zu fördern. Varianten des Ursprungsmythos können dann verwendet werden, um Angst zu schüren und die Akzeptanz von Impfstoffen zu erhöhen, und die „zufällige“ Enthüllung der geheimen GoF-Formel könnte verwendet werden, um technologische Errungenschaften zu verkünden (und möglicherweise auch, um künftige Sündenböcke und Verwirrung zu schaffen, falls dies erforderlich ist).
Absichtliche Verbreitung von nicht-infektiösen Klonen:
- Wenn wir die bereits vorhandene Endemie außer Acht lassen, sind nur einige funktionelle Änderungen an einem bereits vorhandenen lokalisierten Virus (z. B. von einer Fledermaus) erforderlich, das entweder bekannt ist oder neu entdeckt wurde. Da es von einer Fledermaus stammt, wäre es unmöglich, eine GoF-Version für den Menschen zu entwerfen, so dass Tödlichkeit und Übertragbarkeit nicht „eingebaut“ werden könnten. Es wird nicht „viral“ werden, weil es nicht ansteckend ist und keine Pandemie auslösen könnte. Man könnte es jedoch so klonen, dass es neuartige und tödlich aussehende Merkmale enthält, aber abgesehen davon ist es ziemlich egal, welche funktionellen Merkmale es hat, denn die Symptome werden sich nicht von denen anderer Atemwegsviren unterscheiden. Man müsste dann nur diese Klone künstlich verbreiten, damit es wie ein Virus aussieht, das sich ausbreitet. Auch wenn es nicht tödlicher ist als andere Coronaviren, könnte es dennoch Menschen vergiften und eine gewisse Morbidität verursachen. Sie würden dann einen Impfstoff erfinden und einen Test entwickeln, um die mit dem „Virus“ verbundene DNA nachzuweisen. Dies ist, in Kurzform, die Hypothese von JJ Couey [hier]16. Wir gehen davon aus, dass diese Hypothese teilweise durch den Videovortrag von Dr. James Giordano inspiriert ist.
Für beide Szenarien relevant, aber bisher als Faktor bei den Ereignissen der letzten Jahre weitgehend ignoriert, ist die bekannte und weithin erforschte Neigung von chronischem Stress, sich als körperliche Krankheit zu manifestieren. Die Covid-Ära war schließlich durch die nachhaltigste und ausgeklügeltste und koordinierteste Propagandakampagne gekennzeichnet, die jemals über die menschliche Bevölkerung verhängt wurde und speziell darauf abzielte, Angst und Furcht zu schüren. In seinem Videovortrag geht Giordano speziell auf diesen Aspekt ein, wie eine Pandemie erzeugt werden könnte17.
Es sei darauf hingewiesen, dass die Theorie des „nicht ansteckenden Klons“ nicht zu einer sich selbst erhaltenden globalen Pandemie führen würde, sondern eher mit lokalisierten chemischen Angriffen vergleichbar wäre, bei denen Menschen einen über die Luft übertragenen Krankheitserreger einatmen, ihn aber nicht weitergeben würden.
In Anbetracht der operativen Schwierigkeiten, die mit der kontinuierlichen Herstellung und Verteilung von Virusklonen verbunden sind, ist es nach JJ Coueys These möglich, dass die Klone im Jahr 2020 nur an Brennpunkten (NYC und Bergamo, Italien usw.) eingesetzt werden mussten, um eine Scheinpandemie auszulösen, und dass die anschließenden positiven SARS-CoV-2-Fälle eine Kombination aus PCR-Tests, die mit anderen Viren kreuzreagieren und so zu falsch positiven Ergebnissen führen, und/oder natürlichen endemischen SARS-CoV-2-Viren, die mit einer niedrigen Hintergrundrate zirkulieren, waren.
Es gibt noch eine dritte Erklärung, deren Elemente sich mit den vorgenannten überschneiden: dass die Charakterisierung von SARS-CoV-2 als neuartige Entität ein reines Artefakt der virologischen Tests und der genomischen Sequenzierungsmethoden ist. Auf diese Hypothese werden wir hier nicht näher eingehen, freuen uns aber, wenn andere diese Möglichkeit näher untersuchen.
Schlussfolgerung
Das Virus, das als SARS-CoV-2 bekannt geworden ist, scheint aus verschiedenen Blickwinkeln betrachtet gar nicht so neu zu sein. Behauptungen, dass im Jahr 2019 etwas Neues aufgetaucht sei – ob nun durch Gain-of-Function-Experimente oder nicht -, scheinen jeder Grundlage zu entbehren.
Erstens scheint es sich nur um ein weiteres Coronavirus zu handeln, von dem seit den 1980er Jahren mindestens acht weitere ähnliche Viren entdeckt wurden. Darüber hinaus scheint die Letalität des jüngsten Neuzugangs in der Sammlung einen ähnlichen Verlauf zu nehmen wie die der anderen – kurz nach der Entdeckung wird sie überschätzt, um dann später wieder herabgestuft zu werden. Die Zuschreibung der Letalität von Viren leidet unter dem Trugschluss der einzigen Ursache – man geht davon aus, dass das Vorhandensein des Virus ausreicht, um Tod und Krankheit unabhängig von Komorbiditäten und den angewandten (oder nicht angewandten) medizinischen Behandlungen zu erklären. Auch die klinischen Merkmale der mit SARS-CoV-2 assoziierten Erkrankungen scheinen sich kaum von denen anderer Coronaviren und anderer Viren zu unterscheiden, die in jedem Fall häufig als Koinfektion auftreten.
Zweitens erscheinen die viel gepriesenen „besonderen“ strukturellen Merkmale von SARS-CoV-2 bei näherer Betrachtung gar nicht so ungewöhnlich. Ähnliche Merkmale wurden auch bei anderen scheinbar unauffälligen Viren festgestellt. Selbst die gp120-HIV-Inserts könnten von überall dort stammen, wo diese Sequenz in der Natur und beim Menschen vorkommt.
Drittens ist die Komplexität des Verhältnisses zwischen Genotyp und Phänotyp zu groß und schlecht verstanden, als dass die Pathogenität von Viren durch gezielte Eingriffe jemals zuverlässig erreicht werden könnte. Aufgrund des “Fluchs der Dimensionalität” könnte nur die Zeitskala der natürlichen Evolution die dafür benötigte Zeit und den dafür benötigten Raum liefern. Eine Überprüfung der so genannten „Gain-of-Function“- und „Loss-of-Function“-Experimente lässt nicht darauf schließen, dass bei der Bewältigung dieser unüberwindbaren Barriere große praktische und/oder relevante Fortschritte erzielt wurden.
Viertens kann nicht ausgeschlossen werden, dass SARS-CoV-2 seit langem in verschiedenen Tierreservoiren zu verschiedenen Zeiten endemisch war, aber bisher nicht bemerkt wurde. Die inhärenten Grenzen der Verlässlichkeit von phylogenetischen Analysen und Antikörpertests bedeuten, dass wir, um es mit Poppers Worten auszudrücken, eine unbeweisbare Hypothese haben, dass das Virus oder seine Varianten endemisch waren und es für eine unbekannte Zeit gewesen sind.
Fünftens scheint es, dass die Herstellung von Klonen, die genetisches Material tragen, seit geraumer Zeit weltweit gängige Praxis ist und war. In Anbetracht der obigen Ausführungen scheint die Schaffung und Verbreitung solcher Klone eine viel wahrscheinlichere Erklärung für die rasche Verbreitung einer bestimmten Sequenz auf der ganzen Welt zu sein als die natürliche Verbreitung eines neuartigen Erregers aus einer Klasse von Viren, die bekanntermaßen hochgradig veränderlich ist.
In Anbetracht der obigen Ausführungen würden wir vorschlagen, dass Gain-of-Function eher als Claim-of-Function bezeichnet werden sollte.
Uns bleiben daher vier konkurrierende Hypothesen, die das virale „Signal“ erklären könnten: (i) eine bereits bestehende zoonotische Endemie; (ii) ein natürlicher zoonotischer Akt, der das Virus spontan zur richtigen Zeit am richtigen Ort in Wuhan entstehen ließ; (iii) ein „Gain-of-Function“-Laborleck; oder (iv) die (absichtliche?) Verbreitung nicht-infektiöser Klone, die ein globales virales Signal erzeugt. In Anbetracht der Ergebnisse unserer Analyse sind wir der Ansicht, dass die verfügbaren virologischen und epidemiologischen Beweise weder die Theorie des Laborlecks noch die des Nassmarktes für den Ursprung des Virus hinreichend unterstützen.
Abschließend sei gesagt, dass die obigen Ausführungen weder als Entschuldigung für die Unzulänglichkeiten der Virologie (die in den letzten Jahrzehnten ihren Weg verloren zu haben scheint) noch als ausdrückliche Zustimmung zur Gain-of-Function-Forschung dienen sollen. Ein Großteil der Forschung in diesem Bereich scheint weder ethisch noch nützlich zu sein, und obwohl wir nicht glauben, dass Wissenschaftler Viren mit Pandemiepotenzial herstellen können, ist es möglich, ja sogar wahrscheinlich, dass solche Experimente lokal (aber nicht global) Gefahren bergen. Darüber hinaus tragen, wie wir gesehen haben, die ständige Jagd nach Virensequenzen und die hektische Analyse derjenigen, die für neuartig gehalten werden, dazu bei, den Grundstein für eine ständig ängstliche Menschheit zu legen und diejenigen zu ermächtigen, die der Welt eine „Pandemievorsorge“ aufzwingen wollen, mit all ihren Kontrollmechanismen und dem damit einhergehenden Verlust von Freiheiten.
In diesem Artikel wollen wir nicht diejenigen verteidigen, die solche Forschungen durchgeführt haben, aber wir halten es auch nicht für richtig, diese Forschungen allein zu beschuldigen, wenn die Beweise dafür, dass ein sich ausbreitender Erreger eine Pandemie einer neuen Krankheit verursacht hat, so schwach sind. Es ist durchaus möglich, gleichzeitig zu glauben (wie wir oben dargelegt haben), dass eine solche Forschung unethisch ist und dass sie nicht (zumindest nicht direkt) die “Corona-Pandemie“ verursacht hat, die ein iatrogenes Phänomen war. Wenn das Virus tatsächlich eine Funktion hatte, dann die, die Menschheit zu einem dramatischen Akt der Selbstzerstörung zu verleiten.
Nachtrag
Gibt es Hinweise auf eine Endemie vor der Pandemie? Das Vorhandensein von SARS-CoV-2 in Abwasserproben aus Barcelona wurde in einer Vorabveröffentlichung von Chavarria-Miró et al. im Juni 2020 veröffentlicht. Die Abwasserproben wurden von Dezember 2019 bis Mai 2020 archiviert, wobei positive Proben bereits im März 2019 gemeldet wurden, also volle 12 Monate bevor Covid-19 zur Pandemie erklärt wurde. Als die Vorabdrucke schließlich veröffentlicht wurden, hier, wurde die positive Probe vom März 2019 nicht erwähnt, und die früheste gemeldete Probe stammte vom Januar 2020. Dieser Artikel berichtet über gescheiterte Versuche, die Gründe für diese Wendung herauszufinden, und fasst außerdem zahlreiche Studien, hauptsächlich aus Italien, zusammen, in denen vor der Pandemie positive SARS-CoV-2-Proben gemeldet wurden.
Danksagung
Wir sind Freunden und Kollegen dankbar für Ermutigung, Feedback, Einsicht, Überprüfung und Korrektur.
Viele vermuten, dass es sich bei den Fällen von Nierenversagen im Zusammenhang mit „Covid“ in Wirklichkeit um unerwünschte Wirkungen von Remdesivir handelt, das als vermeintliche Behandlung verabreicht wurde.
MERS-CoV und MERS-CoV-ähnliche Antikörper wurden bei Dromedarkamelen nachgewiesen. Kamele sind im Nahen Osten beheimatet. Die Einzigartigkeit scheint als erklärende Variable im Vordergrund gestanden zu haben, und es ist nicht klar, ob andere Tiere getestet wurden. Wäre das Virus in Schottland entdeckt worden, wären vielleicht wilde Haggis der Hauptverdächtige gewesen?
Die Duesberg Die Hypothese, dass HIV kein AIDS verursacht, weist einige auffällige Parallelen zur Geschichte von SARS-CoV-2 auf. Sie beruht auf der Vorstellung, dass HIV ein Passagiervirus ist, das die AIDS-Krankheit nicht verursacht, die stattdessen durch Drogenkonsum (in Europa) und Unterernährung (in Afrika) verursacht wird.
Skeptiker könnten glauben, dass die Öffentlichkeit niemals Informationen zu sehen bekommt, die sie uns nicht zeigen wollen.
Das von Fledermäusen stammende RaTG13 ist derzeit das phylogenetisch am engsten mit SARS-CoV-2 verwandte Virus.
Wir stimmen zwar nicht mit der Annahme überein, dass die Varianten an sich mehr oder weniger pathogen oder übertragbar waren, aber dies ist eine nützliche Heuristik für die wahrscheinliche Entwicklung eines Coronavirus in einer großen Wirtspopulation (oder die beste, die bisher verfügbar ist).
Wir verwenden hier das Wort „Anforderung“ anstelle von „biologischer Fitness“, weil es besser zu der Tatsache passt, dass es funktionell ein menschliches Ziel erfüllen muss.
Ralph Baric: „Wenn man ein Coronavirus an eine andere Spezies anpasst … kann man die Übertragbarkeit auf den Menschen nicht vorhersagen, indem man einfach einen menschlichen Rezeptor einsetzt und ein Mausmodell verwendet“.
Wenn wir davon ausgehen, dass das Ursprungsvirus von einem anderen Tier stammt, müssen wir uns auch mit der Überwindung der Artengrenze auseinandersetzen.
Bei infizierten Frettchen wurden im Vergleich zu Tieren, die mit dem Wildvirus infiziert waren, Läsionen festgestellt, die eine moderate Meningitis und Enzephalitis mit Lymphozyten im Thalamus zeigten. Es wurden jedoch keine Tests auf bakterielle Meningitis durchgeführt, so dass dieser Nachweis in Verbindung mit dem geringen Stichprobenumfang sehr schwach ist.
Diese Forschung zielt eindeutig auf GoF ab, und wenn man anerkennt, dass eine solche Aktivität von Natur aus kriminell ist, dann sind diese Experimente selbst zumindest unethisch. Wer kann außerdem sagen, dass Loss-of-Function weniger gefährlich ist als Gain-of-Function?
Es ist zu beachten, dass die in diesen Studien verwendeten und in der Literatur ausführlich untersuchten Mausmodelle auf einer künstlichen Expression des h-ACE2-Rezeptors beruhen, die das Expressionsmuster desselben Rezeptors beim Menschen und folglich die mit der Infektion verbundene Immunantwort nicht vollständig widerspiegelt.
Derzeit halten die Beweise dafür nicht einmal einer oberflächlichen Prüfung stand.
Sie wissen es nicht; könnte es von Außerirdischen verursacht worden sein?
Die Amendola Studie ging zurück und untersuchte historische Proben auf Antikörper oder virale RNA und fand positive Ergebnisse aus dem Jahr 2019, wobei Teilsequenzen verwendet wurden, die eine Kreuzreaktivität zuließen, aber keines der Ergebnisse war positiv bei der PCR. Außerdem wurde die Studie durch die Tatsache beeinträchtigt, dass die Proben von Masernpatienten stammten, und es wurde davon ausgegangen, dass es einen einzigen Anti-SARS-CoV-2-Antikörpersatz gibt, obwohl diese Viren schwärmen und sich neu kombinieren.
Es könnte sogar viel einfacher sein – eine Mischung aus etwas, das Menschen krank macht (da „es“ alle Symptome verursachen kann, muss es nicht besonders spezifisch sein) und der DNA, die den PCR-Test zum Leuchten bringt.
Ein Aufsatz mit einigen interessanten Beispielen für diesen so genannten „Nocebo“-Effekt findet sich hier.
1 durch ärztliche Maßnahmen entstandenes